Table of Contents
-
Abstract
-
Case Presentations
-
Introduction
-
Critical Appraisal Of The Literature
-
Etiology
-
Pathophysiology
-
Phenotypic Variation In Sickle Cell Disease
-
Differential Diagnosis
-
Prehospital Care
-
Emergency Department Evaluation
-
History
-
Historical Evaluation Of Pain
-
Opiate Addiction And Drug-Seeking Behavior In Sickle Cell Disease
-
Physical Examination
-
Use Of Vital Sign Abnormalities To Assess Pain Severity
-
Diagnostic Studies
-
Can Laboratory Studies Be Used To Objectively Identify Vaso-Occlusive Crisis?
-
Treatment Of Vaso-Occlusion
-
Triage
-
Opiate Therapy
-
Type Of Opiate – Available Evidence
-
Type Of Opiate – Recommendations And Commentary
-
Non-Opiate Therapies
-
Acetaminophen
-
Antihistamines
-
Non-Steroidal Anti-Inflammatory Drugs – Available Evidence
-
Non-Steroidal Anti-Inflammatory Drugs – Recommendations And Commentary
-
Intravenous Fluid Choice – Available Evidence
-
Intravenous Fluid Choice – Recommendations And Commentary
-
Supplemental Oxygen
-
Incentive Spirometry
-
Controversies And Cutting Edge For Vaso-Occlusive Crisis
-
Steroids
-
Magnesium
-
Ketamine
-
Naloxone
-
Nitric Oxide
-
Disposition For Vaso-Occlusive Crisis
-
Special Circumstances: Other Complications Of Sickle Cell Disease
-
Acute Chest Syndrome
-
Etiology And Pathophysiology For Acute Chest Syndrome
-
Emergency Department Evaluation For Acute Chest Syndrome
-
Treatment For Acute Chest Syndrome
-
Oxygen
-
Antibiotics
-
Transfusion
-
Noninvasive Ventilation
-
Steroids
-
Disposition For Acute Chest Syndrome
-
Stroke
-
Treatment For Stroke
-
Fever
-
Treatment For Fever
-
Splenic Sequestration
-
Treatment For Splenic Sequestration
-
Transient Red Cell Aplasia
-
Treatment For Transient Red Cell Aplasia
-
Ophthalmologic Complications
-
Treatment For Ophthalmologic Complications
-
Priapism
-
Treatment For Priapism
-
Clinical Pathway For Management Of Pain In Sickle Cell Disease
-
Risk Management Pitfalls For Sickle Cell Disease
-
Time- And Cost-Effective Strategies For Patients With Sickle Cell Disease
-
Summary
-
Case Conclusion
-
Tables and Figures
-
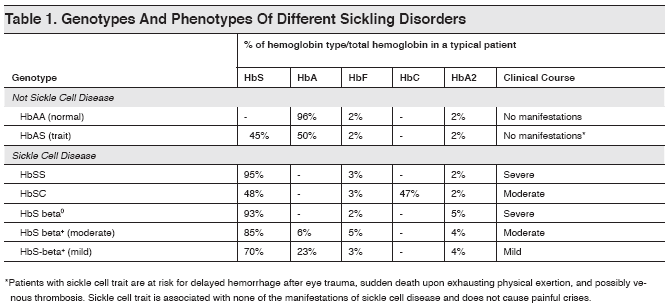
Table 1. Genotypes And Phenotypes Of Different Sickling Disorders
-
Table 2. Acute Complications Of Sickle Cell Disease
-
Table 3. Elements Of The Sickle Cell Disease History
-
Table 4. Opiate Dosing Strategies For Patients > 50 kg With Vaso-Occlusive Crisis
-
Table 5. Non-Opiate Therapies For Vaso[1]Occlusive Crisis
-
Table 6. Differentiating Common Causes Of Acute Anemia In Sickle Cell Disease
-
References
Abstract
Sickle cell disease (SCD) is the most common genetic disease in the US, affecting approximately 100,000 individuals. In SCD, genetically mutated hemoglobin (HbS) forms rigid polymers when deoxygenated, giving red blood cells a characteristic sickled shape. Increased blood viscosity and cell adhesion produce intermittent vaso-occlusion. The vaso-occlusive phenotype of SCD, which is marked by higher hemoglobin, manifests with frequent painful crises and is associated with a higher risk for developing acute chest syndrome. The hemolytic phenotype is characterized by lower baseline levels of hemoglobin and elevated markers of hemolysis. There are no reliable markers of vaso-occlusive crisis (VOC), ie, vital signs and laboratory tests are normal. After intravenous (IV) opiate titration, patient-controlled anesthesia (PCA) pumps are encouraged. Excess IV fluids have been associated with development of atelectasis, a risk factor for acute chest syndrome. Acute chest syndrome has clinical symptoms similar to pneumonia; these patients will develop progressive hypoxemia, acute respiratory distress syndrome, and death if exchange transfusion is not initiated.
Case Presentations
In the middle of a busy evening shift, you encounter a 25-year-old man with a chief complaint of sickle cell crisis. He states that he has had an upper respiratory infection for about a week, followed by progressive pain to his lower back and legs. At home, he has been taking 4-mg tablets of hydromorphone every 3 hours, which reduces his pain score from a 10 to a 9. His vital signs are as follows: heart rate of 77, blood pressure of 115/70 mm Hg, respiratory rate of 14 breaths per minute, and temperature of 36.9°C (98.4°F). He does not appear to be uncomfortable and is sitting in bed using a cellular phone. The nurse has placed a peripheral IV, delivering a 1-L bolus of NS. You get the CBC results, and his hemoglobin is 10.2 mg/dL. You ask yourself several questions:
• Can this patient be having a crisis without a drop in hemoglobin?
• Is there a blood test I can do to confirm that he is truly having a crisis?
• Is the patient addicted to opiates or drug-seeking?
• What fluids should I administer?
• What kind of opiates should I administer?
• Should I administer supplemental oxygen?
• Should I give him IV ketorolac? Are there any other medications that might help?
You are surprised that despite many years of practice you are not sure of the answers and wonder why that is.
Introduction
Sickle cell disease is the most common genetic disease in America,1 affecting approximately 100,000 individuals.2 Since the discovery of SCD 100 years ago, our understanding of the disease has changed dramatically. Research has revealed that the genetic mutations underlying SCD not only result in sickle-shaped red blood cells (RBCs) but also completely alter the rheologic properties of the blood, causing
clinical manifestations in every organ system. Despite major advances in our understanding of the disease, literature to guide clinical decisions in the treatment of SCD is sorely lacking. This issue of Emergency Medicine Practice presents a synopsis of the latest evidence regarding the pathophysiology, diagnosis, and treatment of emergent complications of SCD.
Critical Appraisal Of The Literature
A literature search was performed using PubMed, using the search terms pain, vaso-occlusion, acute chest syndrome, stroke, avascular necrosis, priapism, sepsis, osteomyelitis, transient red cell aplasia, pulmonary hypertension, hyphema, fat embolism, splenic sequestration, hepatic sequestration, hemolysis, and iron overload. A total of 596 articles from 1964 to the present were reviewed. The Cochrane Database was searched for systematic reviews using the key term sickle cell, which identified 35 reviews. Guidelines released by the National Institutes of Health (National Heart, Lung, and Blood Institute) in 2002 and the American Pain Society (APS) in 1999 were also reviewed. Both of these guidelines represent consensus statements and are not systematic, evidence-based guidelines. Using standard evidence-level scales, the majority of clinical evidence in SCD falls into the weaker and moderately-strong categories. There are several reasons for this. First, SCD is a rare disorder, and properly designed clinical trials are often too resource-intensive to perform. Second, SCD research is severely underfunded (cystic fibrosis, a disease that is one-third as common as SCD, receives millions of dollars more in funding each year). A third and final issue is the effect of medical advances on our knowledge of the disease. For example, many observational studies of SCD were performed before the development of the pneumococcal vaccine. This and other advances in preventive medicine may call into question results of earlier observational and interventional trials. When available, recommendations
Risk Management Pitfalls For Sickle Cell Disease
-
“I thought it was just a pain crisis.”
Always consider non-SCD-related conditions in your differential. For example, for right lower quadrant pain, consider appendicitis, kidney stone, and gynecological causes before presuming VOC. For chest pain, consider acute coronary syndromes, pulmonary embolism, or pneumothorax before presuming acute chest syndrome.
-
“He’s had this back pain before, so it can’t be anything dangerous.”
Consider epidural abscess and spinal osteomyelitis in the differential of midline back pain, even when fever is absent.
-
“The hemoglobin was low, so I gave blood. I didn’t think this would cause a stroke.”
Never transfuse a patient simply because hemoglobin is low. Elevating the hemoglobin above baseline can cause hyperviscosity, pain, acute chest syndrome, and stroke.
-
“I thought the patient had sickle cell trait, so I withheld pain medicines.”
When it appears that a patient has sickle cell trait on hemoglobin electrophoresis, make sure the patient did not receive a transfusion within 90 days of having the test performed. Recent transfusion renders the electrophoresis useless for diagnostic purposes.
-
“The patient doesn’t have SCD, so I never thought to check intraocular pressure for such minor eye trauma.”
In cases of direct eye trauma, patients with SCD and sickle cell trait should be treated the same way. Many patients do not know that they carry the trait or will fail to mention it unless prompted. Both SCD and sickle cell trait increase the risk for catastrophic ophthalmologic complications after blunt eye trauma, even if hyphema is not apparent.
-
“I gave the patient a prescription for iron because she was anemic. I didn’t realize this would cause liver problems.”
Never prescribe iron for patients with SCD. These patients are usually iron-overloaded.
-
“The creatinine was normal. How was I supposed to know the patient had kidney dysfunction?”
Assume that all patients with SCD have some degree of renal dysfunction, even if the creatinine level is normal. Supranormal proximal tubule function creates falsely low creatinine in this patient population. Take this into consideration when prescribing NSAIDs and when ordering imaging studies with IV contrast.
-
“I didn’t know that a few liters of normal saline could be harmful.”
Using bolus normal saline to treat sickle cell crisis presents several problems. Excess IV fluid can result in atelectasis, which may precipitate acute chest syndrome. Large amounts of normal saline can produce a hyperchloremic metabolic acidosis, which may promote sickling.
-
“I thought the patient was just faking because he had normal vital signs.”
Most patients with VOC will not exhibit vital sign abnormalities.
-
“The patient wasn’t black, and I thought SCD only occurs in people of African descent.”
Sickle cell disease has been described in all races and should no longer be considered exclusive to black persons.
Tables and Figures
References
Evidence-based medicine requires a critical appraisal of the literature based upon study methodology
and number of subjects. Not all references are equally robust. The findings of a large, prospective, randomized, and blinded trial should carry more weight than a case report.
The most informative references cited in this paper, as determined by the author, are noted by an asterisk (*) next to the number of the reference.
-
Therrell BL, Hannon WH. National evaluation of US newborn screening system components. Ment Retard Dev Disabil Res Rev. 2006;12:236-245.
-
Hassell KL. Population estimates of sickle cell disease in the U.S. Am J Prev Med. 2010;38:S512-S521.
-
Herrick JB. Peculiar elongated and sickle-shaped red blood corpuscles in a case of severe anemia. Archives of Internal Medicine. 6 (5):517–521. doi:10.1001/archinte.1910. http://archinte.ama-assn.org/cgi/content/summary/VI/5/517.
-
Vekilov PG. Sickle-cell haemoglobin polymerization: is it the primary pathogenic event of sickle-cell anaemia? Br J Haematol. 2007;139:173-184.
-
Villagra J, Shiva S, Hunter LA, et al. Platelet activation in patients with sickle disease, hemolysis-associated pulmonary hypertension, and nitric oxide scavenging by cell-free hemoglobin. Blood. 2007;110:2166-72.
-
Sugihara K, Sugihara T, Mohandas N, et al. Thrombospondin mediates adherence of CD36+ sickle reticulocytes to endothelial cells. Blood.1992;80:2634-2642.
-
Belcher JD, Mahaseth H, Welch TE, et al. Critical role ofendothelial cell activation in hypoxia-induced vasoocclusion in transgenic sickle mice. Am J Physiol Heart Circ Physiol. 2005;288:H2715-H2725.
-
Belcher JD, Marker PH, Weber JP, et al. Activated monocytes in sickle cell disease: potential role in the activation of vascular endothelium and vaso-occlusion. Blood. 2000;96:2451-2459.
-
Blann AD, Marwah S, Serjeant G, et al. Platelet activation and endothelial cell dysfunction in sickle cell disease is unrelated to reduced antioxidant capacity. Blood. 2003;14:255-259.
-
Canalli AA, Franco-Penteado CF, Saad ST, et al. Increased adhesive properties of neutrophils in sickle cell disease may be reversed by pharmacological nitric oxide donation. Haematologica. 2008;93:605-609.
-
Graido-Gonzalez E, Doherty JC, Bergreen EW, et al. Plasma endothelin-1, cytokine, and prostaglandin E2 levels in sickle cell disease and acute vaso-occlusive sickle crisis. Blood.1998;92:2551-2555.
-
Brun M, Bourdoulous S, Couraud PO, et al. Hydroxyurea downregulates endothelin-1 gene expression and upregulates ICAM-1 gene expression in cultured human endothelial cells. Pharmacogenomics J. 2003;3:215-226.
-
Conran N, Fattori A, Saad ST, et al.. Increased levels of soluble ICAM-1 in the plasma of sickle cell patients are reversed by hydroxyurea. Am J Hematol. 2004;76:343-347.
-
Duits AJ, Pieters RC, Saleh AW, et al. Enhanced levels of soluble VCAM-1 in sickle cell patients and their specific increment during vasoocclusive crisis. Clin Immunol Immunopathol. 1996;81:96-98.
-
Duits AJ, Rojer RA, van Endt T, et al. Erythropoiesis and serum sVCAM-1 levels in adults with sickle cell disease. Ann Hematol. 2003;82:171-174.
-
Glassberg J, Spivey JF, Strunk R, et al. Painful episodes in children with sickle cell disease and asthma are temporally associated with respiratory symptoms. J Pediatr Hematol Oncol. 2006;28:481-485.
-
Boyd JH, Macklin EA, Strunk RC, et al. Asthma is associated with increased mortality in individuals with sickle cell anemia. Haematologica. 2007;92:1115-1118.
-
Boyd JH, Macklin EA, Strunk RC, et al. Asthma is associated with acute chest syndrome and pain in children with sickle cell anemia. Blood. 2006;108:2923-2927.
-
Gladwin MT, Vichinsky E. Pulmonary complications of sickle cell disease. N Engl J Med. 2008;359:2254-2265.
-
* Platt OS, Brambilla DJ, Rosse WF, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med.1994;330:1639-1644.
-
* Smith WR, Penberthy LT, Bovbjerg VE, et al. Daily assessment of pain in adults with sickle cell disease. Ann Intern Med. 2008;148:94-101.
-
* Platt OS, Thorington BD, Brambilla DJ, et al. Pain in sickle cell disease. Rates and risk factors. N Engl J Med. 1991;325:11-16.
-
* McClish DK, Smith WR, Dahman BA, et al. Pain site frequency and location in sickle cell disease: the PiSCES project. Pain. 2009;145:246-251.
-
Jacob E, Mueller BU. Pain experience of children with sickle cell disease who had prolonged hospitalizations for acute painful episodes. Pain Med. 2008;9:13-21.
-
Sidman JD, Fry TL. Exacerbation of sickle cell disease by obstructive sleep apnea. Arch Otolaryngol Head Neck Surg. 1988;114:916-917.
-
Jones S, Duncan ER, Thomas N, et al. Windy weather and low humidity are associated with an increased number of hospital admissions for acute pain and sickle cell disease in an urban environment with a maritime temperate climate. Br J Haematol. 2005;131:530-533.
-
Vichinsky, E. Overview of the clinical manifestations of sickle cell disease. Available online at www.utdol.com. Accessed 6/28/2011
-
Bijur PE, Latimer CT, Gallagher EJ. Validation of a verbally administered numerical rating scale of acute pain for use in the emergency department. Acad Emerg Med. 2003;10:390-392.
-
Gallagher EJ, Liebman M, Bijur PE. Prospective validation of clinically important changes in pain severity measured on a visual analog scale. Ann Emerg Med. 2001;38:633-638.
-
Garra G, Singer AJ, Taira BR, et al. Validation of the Wong-Baker FACES Pain Rating Scale in pediatric emergency department patients. Acad Emerg Med. 2010;17:50-54.
-
Solomon LR. Treatment and prevention of pain due to vaso-occlusive crises in adults with sickle cell disease: an educational void. Blood. 2008;111:997-1003.
-
Elander J, Lusher J, Bevan D, et al. Pain management and symptoms of substance dependence among patients with sickle cell disease. Soc Sci Med. 2003;57:1683-1696.
-
* Aisiku IP, Smith WR, McClish DK, et al. Comparisons of high versus low emergency department utilizers in sickle cell disease. Ann Emerg Med. 2009;53:5875-5893.
-
* Gladwin MT, Kato GJ, Weiner D, et al. Nitric oxide for inhalation in the acute treatment of sickle cell pain crisis: a randomized controlled trial. JAMA. 2011;305:893-902.
-
* Bartolucci P, El Murr T, Roudot-Thoraval F, et al. A randomized, controlled clinical trial of ketoprofen for sickle-cell disease vaso-occlusive crises in adults. Blood. 2009;114:3742-3747.
-
Ernst AA, Weiss SJ, Johnson WD, et al. Blood pressure in acute vaso-occlusive crises of sickle cell disease. South Med J. 2000;93:590-592.
-
Britto MC, Freire EF, Bezerra PG, et al. Low income as a protective factor against asthma in children and adolescents treated via the Brazilian Unified Health System. J Bras Pneumol. 2008;34:251-255.
-
Chapman JI, El-Shammaa EN, Bonsu BK. The utility of screening laboratory studies in pediatric patients with sickle cell pain episodes. Am J Emerg Med. 2004;22:258-263.
-
Ballas SK, Files B, Luchtman-Jones L, et al. Safety of purified poloxamer 188 in sickle cell disease: phase I study of a non-ionic surfactant in the management of acute chest syndrome. Hemoglobin. 2004;28:85-102.
-
Bouchair N, Manigne P, Kanfer A, et al. Prevention of sickle cell crises with multiple phlebotomies. Arch Pediatr. 2000;7:249-255.
-
Serjeant GR, Petch MC, Serjeant BE. The in vivo sickle phenomenon: a reappraisal. J Lab Clin Med. 1973;81:850-856.
-
Embury SH. The not-so-simple process of sickle cell vasoocclusion. Microcirculation. 2004;11:101-113.
-
Mohammed FA, Mahdi N, Sater MA, et al. The relation of C-reactive protein to vasoocclusive crisis in children with sickle cell disease. Blood Cells Mol Dis. 2010;45:293-296.
-
Standards for the care of adults with sickle cell disease in the UK. 2008. available online http://www.nhlbi.nih.gov/guidelines/scd/index.htm. Accessed 6/28/2011.
-
Benjamin L, Dampier CD, Jacox AK, et al. Guideline for the management of acute and chronic pain in sickle cell disease. Glenview, IL: American Pain Society. 1999.
-
National Institutes of Health. The Management of Sickle Cell Disease. 4th ed. Bethesda, MD: National Heart, Lung, and Blood Institute; 2002:59-74. NIH Publication 02-2117.
-
Robieux IC, Kellner JD, Coppes MJ, et al. Analgesia in children with sickle cell crisis: comparison of intermittent opioids vs. continuous intravenous infusion of morphine and placebo-controlled study of oxygen inhalation. Pediatr Hematol Oncol.1992;9:317-326.
-
Telfer P, Criddle J, Sandell J, et al. Intranasal diamorphine for acute sickle cell pain. Arch Dis Child. 2009;94:979-980.
-
* van Beers EJ, van Tuijn CF, Nieuwkerk PT, et al. Patient-controlled analgesia versus continuous infusion of morphine during vaso-occlusive crisis in sickle cell disease, a randomized controlled trial. Am J Hematol. 2007;82:955-960.
-
* Gonzalez ER, Bahal N, Hansen LA, et al. Intermittent injection vs patient-controlled analgesia for sickle cell crisis pain. Comparison in patients in the emergency department. Arch Intern Med. 1991;151:1373-1378.
-
Jacobson SJ, Kopecky EA, Joshi P, et al. Randomised trial of oral morphine for painful episodes of sickle-cell disease in children. Lancet. 1997;350:1358-1361.
-
Simckes AM, Chen SS, Osorio AV, et al. Ketorolac-induced irreversible renal failure in sickle cell disease: a case report. Pediatr Nephrol. 1999;13:63-67.
-
Schaller S, Kaplan BS. Acute nonoliguric renal failure in children associated with nonsteroidal antiinflammatory agents. Pediatr Emerg Care. 1998;14:416-418.
-
* Hardwick WE, Jr., Givens TG, Monroe KW, et al. Effect of ketorolac in pediatric sickle cell vaso-occlusive pain crisis. Pediatr Emerg Care. 1999;15:179-182.
-
* Wright SW, Norris RL, Mitchell TR. Ketorolac for sickle cell vaso-occlusive crisis pain in the emergency department: lack of a narcotic-sparing effect. Ann Emerg Med. 1992;21:925-928.
-
*Perlin E, Finke H, Castro O, et al. Enhancement of pain control with ketorolac tromethamine in patients with sickle cell vaso-occlusive crisis. Am J Hematol. 1994;46:43-47.
-
* Dunlop R, Bennett KCLB. Pain management for sickle cell disease in children and adults. Cochrane Database of Systematic Reviews. 2006;2:Art. No. CD003350. DOI: 10.1002/1465 1858, pub 2.
-
Guy RB, Gavrilis PK, Rothenberg SP. In vitro and in vivo effect of hypotonic saline on the sickling phenomenon. Am J Med Sci. 1973;266:267-277.
-
* Clark MR, Guatelli JC, Mohandas N, et al. Influence of red cell water content on the morphology of sickling. Blood. 1980;55:823-830.
-
* Jenkins MT, Jones RF, Wilson B, et al. Congestive atelectasis; a complication of the intravenous infusion of fluids. Trans Meet Am Surg Assoc Am Surg Assoc. 1950;68:7-27.
-
* Vichinsky EP, Styles LA, Colangelo LH, et al. Acute chest syndrome in sickle cell disease: clinical presentation and course. Cooperative Study of Sickle Cell Disease. Blood. 1997;89:1787-1792.
-
Embury SH, Garcia JF, Mohandas N, et al. Effects of oxygen inhalation on endogenous erythropoietin kinetics, erythropoiesis, and properties of blood cells in sickle-cell anemia. N Engl J Med.1984;311:291-295.
-
* Lane PK, Embury SH, Toy PT. Oxygen-induced marrow red cell hypoplasia leading to transfusion in sickle painful crisis. Am J Hematol.1988;27:67-68.
-
* Bellet PS, Kalinyak KA, Shukla R, et al. Incentive spirometry to prevent acute pulmonary complications in sickle cell diseases. N Engl J Med.1995;333:699-703
-
Bryant-Stephens T, Kurian C, Guo R, et al. Impact of a household environmental intervention delivered by lay health workers on asthma symptom control in urban, disadvantaged children with asthma. Am J Public Health. 2009;99 Suppl 3:S657-S665.6
-
Zempsky WT, Loiselle KA, Corsi JM, et al. Use of low-dose ketamine infusion for pediatric patients with sickle cell disease-related pain: a case series. Clin J Pain. 2010;26:163-167.
-
Koch J, Manworren R, Clark L, et al. Pilot study of continuous co-infusion of morphine and naloxone in children with sickle cell pain crisis. Am J Hematol. 2008;83:728-731.
-
* Benjamin LJ, Swinson GI, Nagel RL. Sickle cell anemia day hospital: an approach for the management of uncomplicated painful crises. Blood. 2000;95:1130-1136.
-
* Vichinsky EP, Neumayr LD, Earles AN, et al. Causes and outcomes of the acute chest syndrome in sickle cell disease. National Acute Chest Syndrome Study Group. N Engl J Med. 2000;342:1855-1865.
-
Emre U, Miller ST, Rao SP, et al. Alveolar-arterial oxygen gradient in acute chest syndrome of sickle cell disease. J Pediatr.1993;123:272-275.
-
Emre U, Miller ST, Gutierez M, et al. Effect of transfusion in acute chest syndrome of sickle cell disease. J Pediatr. 1995;127:901-904.
-
Liem RI, O’Gorman MR, Brown DL. Effect of red cell exchange transfusion on plasma levels of inflammatory mediators in sickle cell patients with acute chest syndrome. Am J Hematol. 2004;76:19-25.
-
Turner JM, Kaplan JB, Cohen HW, et al. Exchange versus simple transfusion for acute chest syndrome in sickle cell anemia adults. Transfusion. 2009;49:863-868.
-
* Bernini JC, Rogers ZR, Sandler ES, et al. Beneficial effect of intravenous dexamethasone in children with mild to moderately severe acute chest syndrome complicating sickle cell disease. Blood.1998;92:3082-3089.
-
Strouse JJ, Takemoto CM, Keefer JR, et al. Corticosteroids and increased risk of readmission after acute chest syndrome in children with sickle cell disease. Pediatr Blood Cancer. 2008;50:1006-1012.
-
Leikin SL, Gallagher D, Kinney TR, et al. Mortality in children and adolescents with sickle cell disease. Cooperative Study of Sickle Cell Disease. Pediatrics. 1989;84:500-508.
-
Fullerton HJ, Wu YW, Zhao S, et al. Risk of stroke in children: ethnic and gender disparities. Neurology. 2003;61:189-194.
-
Ohene-Frempong K, Weiner SJ, Sleeper LA, et al. Cerebrovascular accidents in sickle cell disease: rates and risk factors. Blood. 1998;91:288-294.
-
Hulbert ML, Scothorn DJ, Panepinto JA, et al. Exchange blood transfusion compared with simple transfusion for first overt stroke is associated with a lower risk of subsequent stroke: a retrospective cohort study of 137 children with sickle cell anemia. J Pediatr. 2006;149:710-712.
-
Adams RJ, McKie VC, Hsu L, et al. Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasonography. N Engl J Med. 1998;339:5-11.
-
Quinn CT, Rogers ZR, McCavit TL, et al. Improved survival of children and adolescents with sickle cell disease. Blood. 2010;115:3447-3452.
-
Hord J, Byrd R, Stowe L, et al. Streptococcus pneumoniae sepsis and meningitis during the penicillin prophylaxis era in children with sickle cell disease. J Pediatr Hematol Oncol. 2002;24:470-472.
-
Neonato MG, Guilloud-Bataille M, Beauvais P, et al. Acute clinical events in 299 homozygous sickle cell patients living in France. French Study Group on Sickle Cell Disease. Eur J Haematol. 2000;65:155-164.
-
Emond AM, Collis R, Darvill D, Higgs DR, Maude GH, Serjeant GR. Acute splenic sequestration in homozygous sickle cell disease: natural history and management. J Pediatr. 1985;107:201-206.
-
Airede AI. Acute splenic sequestration in a five-week-old infant with sickle cell disease. J Pediatr. 1992;120:160.
-
Aslam AF, Aslam AK, Dipillo F. Fatal splenic sequestration crisis with multiorgan failure in an adult woman with sickle cell-beta+ thalassemia. Am J Med Sci. 2005;329:141-143.
-
Hutchins KD, Ballas SK, Phatak D, et al. Sudden unexpected death in a patient with splenic sequestration and sickle cell-beta+-thalassemia syndrome. J Forensic Sci. 2001;46:412-414.
-
Koduri PR. Acute splenic sequestration crisis in adults with sickle cell anemia. Am J Hematol. 2007;82:174-5.
-
Koduri PR, Agbemadzo B, Nathan S. Hemoglobin S-C disease revisited: clinical study of 106 adults. Am J Hematol. 2001;68:298-300.
-
Koduri PR, Kovarik P. Acute splenic sequestration crisis in an adult with sickle beta-thalassemia. Ann Hematol. 2006;85:633-635.
-
Koduri PR, Nathan S. Acute splenic sequestration crisis in adults with hemoglobin S-C disease: a report of nine cases.Ann Hematol. 2006;85:239-243.
-
Manci EA, Culberson DE, Yang YM, et al. Causes of death in sickle cell disease: an autopsy study. Br J Haematol. 2003;123:359-365.
-
Fernandes AP, Januario JN, Cangussu CB, et al. Mortality of children with sickle cell disease: a population study. J Pediatr (Rio J). 2010;86:279-284.
-
Smith-Whitley K, Zhao H, Hodinka RL, et al. Epidemiology of human parvovirus B19 in children with sickle cell disease. Blood. 2004;103:422-427.
-
Mallouh AA, Qudah A. Acute splenic sequestration together with aplastic crisis caused by human parvovirus B19 in patients with sickle cell disease. J Pediatr. 1993;122:593-595.
-
Goldstein AR, Anderson MJ, Serjeant GR. Parvovirus associated aplastic crisis in homozygous sickle cell disease. Arch Dis Child. 1987;62:585-588.
-
Kurtzman G, Frickhofen N, Kimball J, et al. Pure red-cell aplasia of 10 years’ duration due to persistent parvovirus B19 infection and its cure with immunoglobulin therapy. N Engl J Med. 1989;321:519-523.
-
Kurtzman GJ, Cohen B, Meyers P, et al. Persistent B19 parvovirus infection as a cause of severe chronic anaemia in children with acute lymphocytic leukaemia. Lancet. 1988;2:1159-1162.
-
Frickhofen N, Abkowitz JL, Safford M, et al. Persistent B19 parvovirus infection in patients infected with human immunodeficiency virus type 1 (HIV-1): a treatable cause of anemia in AIDS. Ann Intern Med. 1990;113:926-933.
-
Miller E, Fairley CK, Cohen BJ, et al. Immediate and long term outcome of human parvovirus B19 infection in pregnancy. Br J Obstet Gynaecol. 1998;105:174-178.
-
Mowatt L, Chambers C. Ocular morbidity of traumatic hyphema in a Jamaican hospital. Eur J Ophthalmol. 2010;20:584-589.
-
Nasrullah A, Kerr NC. Sickle cell trait as a risk factor for secondary hemorrhage in children with traumatic hyphema. Am J Ophthalmol. 1997;123:783-790.
-
Coats DK, Paysse EA, Kong J. Unrecognized microscopic hyphema masquerading as a closed head injury. Pediatrics. 1998;102:652-654.
-
Walton W, Von Hagen S, Grigorian R, et al. Management of traumatic hyphema. Surv Ophthalmol. 2002;47:297-334.
-
Pakalnis VA, Rustgi AK, Stefansson E, et al. The effect of timolol on anterior-chamber oxygenation. Ann Ophthalmol. 1987;19:298-300.
-
Gharaibeh A, Savage HI, Scherer RW, et al. Medical interventions for traumatic hyphema. Cochrane Database Syst Rev. 2011:CD005431.
-
Mantadakis E, Cavender JD, Rogers ZR, et al. Prevalence of priapism in children and adolescents with sickle cell anemia. J Pediatr Hematol Oncol. 1999;21:518-522.
-
Karayalcin G, Imran M, Rosner F. Priapism in sickle cell disease: report of five cases. Am J Med Sci. 1972;264:289-293.
-
Rifkind S, Waisman J, Thompson R, et al. RBC exchange pheresis for priapism in sickle cell disease. JAMA. 1979;242:2317-2318.
-
Seeler RA. Intensive transfusion therapy for priapism in boys with sickle cell anemia. J Urol. 1973;110:360-363.
-
Walker EM Jr, Mitchum EN, Rous SN, et al. Automated erythrocytopheresis for relief of priapism in sickle cell hemoglobinopathies. J Urol. 1983;130:912-916.
-
Abboud MR, Musallam KM. Sickle cell disease at the dawn of the molecular era. Hemoglobin. 2009;33 Suppl 1:S93-S106.
-
Rackoff WR, Ohene-Frempong K, Month S, et al. Neurologic events after partial exchange transfusion for priapism in sickle cell disease. J Pediatr. 1992;120:882-885.
-
Siegel JF, Rich MA, Brock WA. Association of sickle cell disease, priapism, exchange transfusion and neurological events: ASPEN syndrome. J Urol.1993;150:1480-1482.
-
McHardy P, McDonnell C, Lorenzo AJ, et al. Management of priapism in a child with sickle cell anemia; successful outcome using epidural analgesia. Can J Anaesth. 2007;54:642-645.
-
Labat F, Dubousset AM, Baujard C, et al. Epidural analgesia in a child with sickle cell disease complicated by acute abdominal pain and priapism. Br J Anaesth. 2001;87:935-936








 678-366-7933
678-366-7933