
|
|
Cocaine (benzoylmethylecgonine) is found in the leaves of the Erythroxylon coca bush in the Andean region of South America. The leaves are chewed for traditional, medicinal, and religious purposes even today; in the mid 19th century, cocaine hydrochloride (the active alkaloid) was extracted from coca leaves and recognized as a powerful local anesthetic and vasoconstrictor. subsequently, it was commonly used as an anesthetic for many surgical procedures. Its popularity increased and it became commercially available during the early 20th century in many tonics and was even an active ingredient in Coca-Cola™ before being recognized as a dangerously addictive drug. Except for limited medical use, it was banned in the U.S. in 1914 and has remained banned despite frequent resurgences within segments of the population who have sought it out for its mind altering effects.1
Cocaine hydrochloride is the crystalline salt form of cocaine that can either be injected or nasally insufflated. On the street, cocaine is sold under the monikers "coke," "blow," and "snow."2 The drug's most recent epidemic commenced in the 1970s when it became a drug of popular abuse. In the 1980s, the cocaine alkaloid derivatives, "freebase" and "crack," invaded popular urban culture. Their use became attractive due to their inexpensive cost, relative ease of use, and intense and immediate effects.3
From the 1970s through the 1990s, the use of cocaine reached epidemic proportions as recorded by the Substance Abuse And Mental Health Administration (SAMHSA) in their annual National Survey on Drug Use and Health (NSDUH) reports. These reports showed that the percentage of young adults aged 18 to 25 who had ever used cocaine was below 1% during the mid-1960s. However, the rate rose steadily throughout the 1970s and early 1980s, reaching 17.9% in 1984. By 1996, the rate dropped to 10.1%, but it climbed to 15.4% in 2002.4
During the height of the 1980s cocaine epidemic, 57% of patients presenting with cocaine-related chest pain were admitted to the hospital to exclude myocardial infarction (MI); 5.7% sustained an MI as measured by CK/MB.5 This practice cost the health care system an estimated $83 million dollars per year,6 and spurred the need for better understanding of the pathophysiology of cocaine intoxication and treatment options. This issue of Emergency Medicine Practice discusses the general management of cocaine-associated emergencies. Additionally, it will make evidence-based recommendations for the treatment and disposition of these patients.
It's another busy Saturday night in the ED when the nurse hands you the triage sheet of a 36-year-old male complaining of chest pain. The vital signs are significant for tachycardia and hypertension. Just then, you look up to see a commotion in triage as a handcuffed patient is escorted in by the police. He appears agitated and is wearing a bloody shirt, reeking of sweat and beer. The police inform you that the patient was involved in a fight and was found driving around with a large amount of cocaine on his person. They insist that he only started complaining of chest pain to avoid being locked up. You look down at the ECG which shows sinus tachycardia with lateral T wave inversions. The patient asks, "Am I having a heart attack?" while the officer asks, "Can I take this guy to jail?" You answer, "No," to both of them, though you wonder if you answered too quickly. Thirty minutes later, an ALS crew brings in a patient found in a college dormitory. He is well dressed and obtunded. His friend reports that the patient had been sniffing cocaine when he complained of a headache and suffered what may have been a seizure. It appears that half of the dormitory is in the waiting room anxiously demanding to know, "What's going to happen to Fred?"
A MEDLINE search was conducted using the keywords cocaine, crack, acute coronary syndrome, acute myocardial infarction, chest pain, cerebral vascular accident, stroke, seizure, and renal failure. This search produced several hundred articles, 190 of which were selected for inclusion in this review. While much of this literature is comprised of retrospective analyses, many articles on the management of the cardiovascular manifestations of cocaine intoxication involve recent large prospective studies. The American Heart Association (AHA) Guidelines 2000 for Cardiovascular Resuscitation of Emergency Cardiovascular Care are incorporated in this review. This was the most comprehensive source for evidence-based treatment recommendations of cocaine-associated myocardial ischemia. A review of the Cochrane database and the National Guideline Clearinghouse provided information on substance abuse in general, but there were no recommendations for management of acute cocaine toxicity. The literature regarding the prevalence of cocaine in society and mentions in emergency department literature was obtained using the database of the Drug Abuse Warning Network (http://Dawninfo.samhsa.gov/) and the National Institute on Drug Abuse (http://www.nida.nih.gov/).
In the early 1990s, cocaine ranked among the leading causes of death for young adults in New York City, where cocaine metabolites were found in over 25% of fatal injury autopsies.7 Cocaine was also found in 9-13% of homicide victims.89 Today, cocaine is second only to alcohol as a toxin-related cause of ED visits.10 The National Institute of Drug Abuse (NIDA) identifies adult males 18-25 years of age as having the highest rate of cocaine use,11 though intake is not limited to this age group and has spread across all demographics. According to NSDUH, 2.4% of teenagers in the age range of 12 to 17 used cocaine in 2004. People seeking treatment for cocaine use surged from 276,000 in 2003 to 466,000 in 2004. This trend toward increased cocaine prevalence in society is reflected in the numbers presenting to EDs with cocaine-related complaints. The Drug Abuse Warning Network (DAWN) report found 199,198 mentions of cocaine use in ED visits in 2002. This represents a 39% increase from ED mentions in 1994 while total ED visits rose by only 15%.11 In 2005, DAWN estimated that there were 448,481 cocaine-related ED visits. Though there was increased surveillance and DAWN estimates represent a limited sample of ED visits this demonstrates that cocaine accounts for roughly one in three drug-related ED visits.10As cocaine affects vascular tone, it has an extensive physiologic influence on all of the body's organs. It is no surprise that cardiopulmonary complaints after cocaine use are the most common. The sum total of cardiopulmonary complaints may account for up to 56.2% of cocaine-related complaints in the ED. Chest pain was reported by 40% of patients presenting to EDs with recent cocaine use. Neurological symptoms (39.1%) are the second leading cause of cocainerelated complaints, followed by psychiatric (35.6%), general/constitutional (23.5%), gastrointestinal (13.3%), and ophthalmologic/ otorhinolaryngologic (8.2%). 12
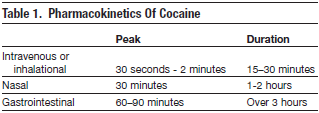
Cocaine hydrochloride is the water-soluble form of cocaine that can be injected, insufflated, or absorbed across all of the body's mucous membranes. "Freebasing" is the home conversion of cocaine salt into a purified form by dissolving and heating it to form a purified alkaloid paste. Crack or "rock" is the preprocessed freebase and is sold inexpensively as small crystallized rocks. Freebase and crack are heat stable, a property that allows them to be smoked in pipes or added to tobacco.2 These vapors are fat-soluble and enable rapid absorption across the blood-lung and then the blood-brain barriers.2 On the street, cocaine may often be sold contaminated with other agents such as talc, caffeine, amphetamine, mannitol, lidocaine, and strychnine which may add to the toxicity. Peak onset of action and duration of cocaine toxicity depend on the route of administration (Table 1) .

There are two routes by which cocaine is metabolized. Enzymatic hydrolysis into ecgonine methyl esther (EME) by liver and plasma esterases accounts for 30-50% of cocaine's metabolism. This metabolic pathway is clinically important since patients with relative plasma cholinesterase deficiency appear to be at increased risk of adverse consequences from cocaine toxicity.13 Cocaine is also nonenzymatically hydrolyzed to benzoylecgonine (BE) roughly 40% of the time. BE has lesser vasoconstrictive properties when compared to cocaine; it is the metabolite that is tested in most urine immunoassays. In on-habitual users, cocaine is detected for 48 to 72 hours in the urine; it may be detected for up to three weeks in chronic, high dose cocaine users. 14,15 Approximately 10% of cocaine is also metabolized to norcocaine and ecgonine. Norcocaine is highly vasoconstrictive and produces similar effects as the parent compound.16
Cocaine users often co-ingest ethanol with cocaine. They report that the co-abuse of ethanol and cocaine prolongs the euphorigenic properties of cocaine while minimizing the dysphoria of withdrawal.17 In the presence of ethanol, the metabolite cocaethylene (also called ethylcocaine) is formed via transesterification by liver esterases.18This metabolite has similar pharmacologic properties as cocaine. It is a potent vasoconstrictor and its clinical effects may lead to infarction or sudden death.
Cocaine has local and systemic effects that are derived from both its sodium channel blockade and monoamine reuptake inhibition. The first mechanism is responsible for cocaine's local anesthetic effects. Transient inhibition of sodium flux across cell membranes during depolarization inhibits nerve conduction and causes anesthesia.19 In fast sodium channels in the myocardium, cocaine imparts type I antidysrhythmic properties, resulting in depression of myocardial depolarization and slow conduction. This property prolongs the action potential, widens the QRS complex, and impairs cardiac inotropy.20 Cocaine's second mechanism of action is responsible for the release of catecholamines from central and peripheral stores. 20 This effect is achieved by cocaine's ability to bind to the monoamine reuptake pump of presynaptic neurons and, consequently, to increase synaptic monoamine neurotransmitter (norepinephrine, epinephrine, dopamine, and serotonin) concentrations. This action results in a panoply of central and peripheral symptoms. In the central nervous system (CNS), increased norepinephrine and dopamine concentrations stimulate postsynaptic β-adrenergic receptors causing psychomotor agitation, diaphoresis, mydriasis, and tremor.21 In the periphery, cocaine has direct action on the adrenal gland (release of epinephrine) and on vascular smooth muscle. The overall increase in synaptic and circulating catecholamines which clinically manifest as hypertension, tachycardia, diaphoresis, mydriasis, and hyperthermia are the classic components of the sympathomimetic toxidrome.20,21 The additional symptoms of cocaine toxicity are related to the respective neurotransmitter involved. An increase in dopamine is responsible for the increase in locomotion,22 restlessness, agitation, and seizures as well as for the sense of euphoria, reinforcement, and addiction.23 The effects of cocaine on serotonergic mediated processes are unclear. Serotonin acts on CNS sites that are attributed several psychological effects such as mood, appetite, personality, affect, motor function, sexual activity, sleep induction, and hallucination as well as temperature regulation and vasospasm. Cocaine increases excitatory amino acid levels by enhancing dopamine stimulation of N-methyl-D-aspartate (NMDA) receptors.24 Recent animal models describe an attenuation of cocaine-induced convulsions with excitatory amino acid antagonists.25,26
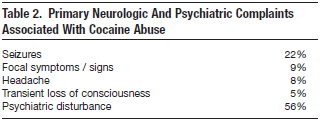
In 1977, Brust and Richter comprehensively described CNS complications from cocaine hydrochloride use including cerebrovascular ischemia and infarction, subarachnoid hemorrhage, intraparenchymal hemorrhage, transient ischemic attack, seizures, cerebral vasculitis, migraine headaches, anterior spinal artery syndrome, movement disorders, and cerebrospinal fluid leakage27-39(Table 2). Similar cerebrovascular complications are found in patients who use crack cocaine, with symptoms occurring immediately or within one hour of use in 64% of patients.28
Cocaine-induced strokes can be ischemic or hemorrhagic and may be caused by both powdered cocaine (hydrochloride) and crack (alkaloidal).28,29 While ischemic and hemorrhagic strokes are equally likely to occur after crack cocaine use, powdered cocaine is four times more likely to cause hemorrhagic strokes.29
Cocaine-induced hemorrhagic strokes are generally either subarachnoid (SAH) or intracerebral.40 Cases of SAH after cocaine use were first reported in 1984.41 SAHs have a higher mortality than intracerebral hemorrhages or ischemic strokes (CVAs) and are mostly found in patients with underlying neurovascular anomalies such as aneurysms.35
Autopsy studies of cocaine-induced intracerebral hemorrhages most often fail to demonstrate specific pathologic cerebral lesions or inflammatory changes. This lack of detectable vascular abnormalities has led experts to believe that a temporal event, such as hypertensive surges and vasospasm, may be the likely mechanism for the hemorrhage.42,43 One of these autopsy studies found the incidence of hypertensive cardiovascular disease to be significantly higher in persons with intracerebral hemorrhage secondary to cocaine than in those with aneurysm rupture secondary to cocaine. These findings suggest that underlying hypertensive cardiovascular disease predisposes cocaine users to intracerebral hemorrhages. This may occur by shifting the limit of cerebral autoregulation to lower blood pressure levels.44 Cocaine may achieve this action via its dopaminergic activity since dopamine is known to lower the upper limit of autoregulation.
Ischemic strokes and transient ischemic attacks (TIAs) after cocaine use are likely a result of cerebral vasoconstriction, hypertension, or thrombi/emboli.30 Experimental data demonstrate that there is a direct relationship between cocaine administration and human cerebral vasoconstriction. Intravenous administration of cocaine (0.2 or 0.4 mg/kg) in healthy, medically and neurologically normal individuals who are occasional cocaine users demonstrated a dose-related cerebral vasoconstriction on magnetic resonance angiograms (MRAs). The degree of vasoconstriction also correlated with frequency of selfreported lifetime cocaine use. The latter suggests a cumulative effect of vasoconstriction in chronic cocaine users.45 Thrombi/emboli potentiated by cocaine directly or via cardiogenic events have been described. 46 However, exact data on ischemic CVAs cannot be ascertained, as ischemic lesions are vulnerable to transform into hemorrhagic lesions.
Cocaine-induced seizures are described as generalized with tonic-clonic features and are reported to occur within minutes (intravenous) to 12 hours after cocaine use.47 Cocaine precipitates seizures in patients with and without seizure disorders by lowering the seizure threshold in multiple ways.48 Animal models have implicated an adrenergic surge of hypertension, hyperthermia, and vasospasm.49 Increased serotonergic neuronal activity has been shown to contribute to cocaine-induced convulsions.50 Cocaine-poisoned mice have a reduced catalase activity which suggests that oxidative stress and inhibition of dopamine may potentiate the neurotoxic effects of cocaine.51 Cocaine also directly increases the concentration of intracellular calcium which prompts cerebral vasoconstriction, exacerbates the catecholaminergic effects of the drug, and facilitates seizures.52 Other studies suggest that habitual use of cocaine sensitizes the brain and has a 'kindling' effect to promote seizures. Kindling refers to animal studies demonstrating that repeated electrical stimulation of subcortical structures in the brain is associated with an increase in seizure susceptibility. 53,54
After cocaine use, migraine-like headaches have been reported. These headaches vary in intensity, quality, location, and time of resolution.47 Interestingly, a case series described three patients with cocaine "withdrawal" headaches that occured several hours after a cocaine binge which then aborted after cocaine or ergotamine administration. The patients in this study may have suffered a depletion of serotonin and experienced migraine-like symptoms that reversed with the administration of cocaine, just as migraines abate with serotonin agonists such as ergots.38
Movement disorders (such as dystonias, choreoathetosis [crack dancing], and akathisia) have been attributed to the dopaminergic effects of cocaine. Patients on neuroleptic medications are particularly predisposed to these extrapyramidal dysfunctions.32,55
Cocaine has a variety of deleterious effects on the heart; these can best be understood by dividing them into acute/intermediate and delayed or chronic effects. Cocaine acts as an acute and direct myocardial depressant. In-vitro animal studies demonstrate that high concentrations of cocaine have a negative inotropic effect on the myocardium. This effect is caused by cocaine's properties as a sodium channel blocker on the myocardial sarcolemma.
Acute coronary artery vasoconstriction after cocaine use is mediated through α-adrenergic stimulation. Experimental human studies of intranasal cocaine during cardiac catheterization have demonstrated diffuse coronary artery narrowing by approximately 13%. This arterial vasoconstriction is markedly worse (29% narrowing) in coronary artery segments already damaged by atherosclerosis.56 Vasoconstriction after cocaine use is similar to that observed after cigarette smoking.57 An angiographic study described how the combination of intranasal cocaine and cigarette smoking narrowed diseased coronary artery segments and increased myocardial oxygen demand to a greater degree than either cocaine or cigarette smoking alone.58 Therefore, chronic cocaine users who are smokers are at greater risk for cocaine-induced vasoconstriction. It has also been demonstrated that while intracoronary administration of phentolamine (an α-adrenergic blocking agent) reversed cocaine-induced vasoconstriction, propranolol (a β-adrenergic antagonist) potentiated this action.59,60
The vascular events associated with cocaine use are not completely explained by the adrenergic stimulation alone. Cocaine-induced thrombosis is a potential cause of delayed ischemia and infarction in patients presenting with cocaine-associated chest pain after the sympathomimetic stimulation has abated. Occasionally, occlusive thrombi are found post mortem in the setting of both normal and diseased coronary arteries.61 The mechanism by which cocaine may induce thrombogenicity is not completely understood. Cocaine impacts the homeostasis between thrombosis and fibrinolysis through direct endothelial damage, platelet aggregation, and fibrin deposition.62 A rabbit model of cocaine toxicity demonstrated direct endothelial injury histologically. These injuries may form the nidus for platelet activation and fibrin deposition as well as atherosclerosis.63 Togna et al noted that cocaine increases platelet membrane aggregation and thromboxane synthesis invitro.64 Additionally, in-vitro studies demonstrate that cocaine alters plasma constituents such as plasminogen activator inhibitor (PAI-1), a von Willebrand factor that regulates thrombus formation.65-67
Cocaine-associated chest pain is reported in patients several weeks after the cessation of cocaine use. Withdrawal from cocaine has been associated with a dopamine-depleted state as well as spontaneous vasospasm and ischemic episodes as monitored on ECGs of patients entering drug treatment programs.68
The exact cause of rhythm disturbances in cocaine toxicity is unclear. However, there are several acute and chronic mechanisms that act independently or concomitantly to produce them. Cocaine's direct sodium channel blockade impedes the heart's conduction system. Canine models of cocaine toxicity confirmed cocaine's Type 1A sodium channel blocking properties. Both a dose-dependant QRS interval prolongation and a sinus cycle length increase are observed. The QRS selectively narrows after the administration of sodium bicarbonate experimentally and clinically.69,70 In addition, cocaine intoxication raises the levels of circulating epinephrine and norepinephrine up to fivefold, which in itself may promote cardiac electrical instability.21 These endogenous catecholamines may also cause myocardial ischemia or infarction, predisposing the heart to dysrhythmias.
Chronic cocaine use causes structural abnormalities that provide a substrate for reentrant dysrhythmias and other conduction disturbances.19 Autopsy reports of cocaine users find atherosclerotic changes and left ventricular hypertrophy.71 Cocaine administered to cholesterol fed rabbits increased the prevalence of atherosclerosis in these animals.63 Other structural cardiac abnormalities described in autopsy reports are myocarditis, contraction band necrosis (hyper-contracted or ruptured cardiac sarcomeres), and cardiomyopathy (in habitual cocaine users). Some of these changes occur in the absence of coronary artery disease.19,72,73 The cause of these chronic changes after cocaine use has not been fully elucidated. An experimental animal study explained some of these changes through cocaine's ability to modify gene expression.74
The first challenge in treating cocaine-associated emergencies is to correctly identify this group of patients. A broad range of conditions may mimic the classic sympathomimetic toxidrome of acute cocaine intoxication. Those conditions that are exactly mimicked include some forms of drug intoxication and endocrine disorders. Other conditions may have many of the signs and symptoms of cocaine intoxication. Many other illnesses have only a few of the manifestations of cocaine toxicity and should not be overlooked (Table 3).

It is important to note that acute cocaine manifestations are governed by the dose, time, and method of use. For instance, some patients present with cocaine-related complaints (such as chest pain) long after the clinically apparent sympathomimetic effects of cocaine resolve. Likewise, symptoms may be masked by other drugs such as alcohol, opioids, or anticholinergic medications. Consequently, emergency physicians should have a high degree of suspicion of recent drug use in all patients. It has been reported that only 13% of patients presenting with chest pain are queried in the ED about recent cocaine use. Furthermore, patients are frequently not forthcoming about their drug use when asked by a physician. One study of three suburban EDs reported a 29% likelihood of testing positive for cocaine in patients aged 18 to 30 presenting with chest pain and 48% for ages 31 to 40 years.75,76 Given these confounders, all fitting patients should be asked about recent cocaine use and a toxicology screen should be considered to help identify these patients if cocaine use is suspected but not volunteered.
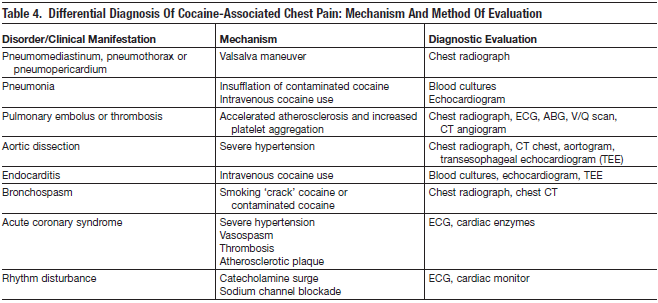
If cocaine intoxication has been revealed in a patient presenting with a given complaint, the evaluation should proceed with a broad differential as this group is at high risk for the same maladies that may mimic cocaine intoxication. For instance, patients presenting with chest pain have to be evaluated not only for cardiac complications of cocaine but also for vascular, pulmonary, and gastrointestinal etiologies. Case reports of cocaine-related aortic rupture and dissection have been described.77 Pneumothorax from Valsalva maneuvers when the drug is inhaled or insufflated is a reported complication of drug use.78 Intravenous cocaine users are at risk for developing upper extremity deep venous thrombosis and endocarditis. 79 The evaluation of cocaine-intoxicated patients should ensue in the same manner as with patients with traditional risk factors for atherosclerotic heart disease and cerebrovascular disease.
Prehospital care for cocaine-intoxicated patients follows general treatment guidelines since there are no studies that are specifically directed for these patients. Patients with suspected acute cocaine intoxication and cocaine-related complaints may require immediate and aggressive anxiolysis with benzodiazepines. Notations of the scene and bystander comments are beneficial in the patient with altered mental status. All patients should be screened in the field with vital signs, cardiac and pulse oximetry monitoring, and supplemental oxygen. Testing of plasma glucose concentration or empiric dextrose is also required for agitated patients. Thiamine may be supplemented as well at this time.
The administration of aspirin to patients with cocaine-associated chest pain has not been studied. However, aspirin has a good safety profile in patients with chest pain and traditional risk factors of atherosclerotic disease and may be safely administered in patients with cocaine-associated chest pain. Clinical studies suggest that sublingual nitroglycerin (SL NTG) is safe for cocaine-related chest pain.130 Patients presenting with cocaine-associated chest pain or with stroke symptoms should be preferentially transported to a center able to perform percutaneous coronary intervention (PCI) or to a stroke center, respectively.
Since cocaine intoxication can be associated with traumatic injury, all patients should be assessed for signs of trauma. Transport to an appropriate trauma facility should follow accordingly.
Most patients presenting with acute cocaine intoxication or cocaine-related complaints exhibit sympathomimetic signs. This may range from a patient who is severely agitated, confused, and combative to one who is calm with all or some of the classic signs of the sympathomimetic toxidrome. In the agitated or altered and confused patient, intravenous access, supplemental oxygen, and cardiac monitoring that began in the field should be continued. The patient should be fully undressed, examined and when necessary secured and restrained.
A full history should be obtained from the patient, with emphasis on cardiovascular and CNS symptomatology to screen for life-threatening cerebrovascular complications of cocaine use.80 EMS and bystanders who brought the patient into the ED should be interviewed as well.
Most patients report the onset of neurological symptoms after using cocaine in three phases: during or immediately after its use (54.5%), within six hours (33.3%), and between six and twelve hours (6.1%). The most common complaints are headache, focal neurological deficits, meningismus, and dysphasia.35
Cocaine intoxication is uncommonly associated with seizures (2.8-8.4%).47,81,82 Similar to CVAs, seizures largely occur within 90 minutes of cocaine usage.54 Most cocaine-induced seizures are reported to be single, generalized, and tonic-clonic. Focal seizures (20%) or multiple seizures are associated with intracerebral pathology. A retrospective cohort study found that patients with epilepsy are more than twice as likely of having cocaine-induced seizures than patients without a history of epilepsy.53 The frequency of seizures in patients with a history of epilepsy was 16.9% while the frequency in patients without a history of epilepsy was 7.9%. In patients with a prior history of epilepsy, 41.7% had focal seizures and 66.7% had multiple seizures. In comparison, only 9.4% of patients without a prior history of epilepsy had focal seizures, and 28.1% had multiple seizures.48
Forty percent of cocaine-related visits to EDs list chest pain as the leading single complaint. Two-thirds of patients report cocaine-associated chest pain within the first three hours. Onset of chest pain corresponds to the route of cocaine administration: intravenous, smoked, then nasally insufflated.83 There is a 24-fold increased risk of MI in the first hour following cocaine use.84 Other frequently occurring cardiac-related complaints at presentation include diaphoresis, palpitations, and dyspnea.12,85A prospective observational cohort study of patients presenting with cocaine-associated chest pain described these patients as young tobacco-smoking men with frequent cocaine use and often no other cardiac risk factors. The authors in this study found no statistical difference in the characteristics of chest pain, location, quality, duration, vital signs, or route of drug administration between those patients who ruled in and those who ruled out for MI. Associated symptoms of chest pain, such as shortness of breath, diaphoresis, palpitations, nausea, vomiting, and syncope, were not predictive of MI.86
Crack cocaine smokers frequently report an array of respiratory complaints after smoking. These symptoms include cough, black sputum, chest pain, shortness of breath, and asthma.87 Status asthmaticus also occurs from nasal insufflation of cocaine.88 There is some suggestion that the increase in asthma severity and mortality is possibly related to the increase in prevalence of crack cocaine use as a precipitating factor in this study population in particular. The authors claim that these results may be extrapolated to the entire U.S.89
Vital signs and the ABCs are the fist step in evaluating these patients. Airway and oxygenation should be assessed and continuously monitored. Thermal upper airway injuries of the tongue, epiglottis, vocal cords, and subglottic areas can occur after smoking cocaine or inhaling the ether used to prepare the alkaloidal form of cocaine.90
A rapid pulse may signify acute cocaine intoxication, dehydration, blood loss, or agitation. The heart should be monitored for rate and dysrhythmias. Frequent blood pressure measurements should be obtained to identify a hypertensive urgency, and discrepancies in bilateral blood pressure measurements may reflect aortic pathology (e.g., aortic dissection).91
Core temperature measurements in patients with an altered sensorium are a necessity. Hyperthermia is the vital sign abnormality that correlates most with fatality in cocaine users. There are numerous case reports of hyperthermia-related deaths with or without rhabdomyolysis after cocaine use.92,93 Cocaine causes hyperthermia in several ways. Cocaine increases heat production through psychomotor agitation via its CNS effects. Cocaine also controls the dopamine-modulated heat-regulatory centers of the hypothalamus.94 Peripherally, cocaine hampers heat dissipation by vasoconstriction of the vasculature. In addition, high ambient temperatures are associated with an increase in mortality from cocaine overdose.95 In a medical examiner surveillance study in New York City, it was found that significantly more deaths were due to cocaine overdoses on hot days than on other days. The mean daily mortality began to increase when maximum temperature equaled or exceeded 31.1 C (88 F). These findings are consistent with data demonstrating that the survival rate of cocaine-poisoned dogs fell from 100% to 57% when ambient temperature was increased from 5 C to 5 C.49
It must be noted that anticholinergic toxicity can also present with altered mental status, tachycardia, and hyperthermia. Clinically, it will differentiate itself from cocaine toxicity by unreactive mydriasis, dry skin and mucosa, absent bowel sounds, and urinary retention on physical examination.
A prompt neurological evaluation is imperative in all intoxicated patients as cocaine is associated with fatal CVAs and poor Glasgow Coma Scores (GCSs).80Assessment of cranial nerve palsies and focal sensory and motor deficits on the neurological examination is important. In a study of patients with ischemic and hemorrhagic cocaine-related CVAs, patients presented awake, alert, or decerebrate. Their average GCS score was 11.35 Signs such as hemiplegia, dysarthria, aphasia, and paresthesias are well described in cocaine-induced CVAs.95 Patients presenting with any of these neurological sequelae have a mortality of 27.3%.35
Auscultation of the chest may reveal dysrythmias, murmurs, and evidence of barotrauma. The Valsalva mechanism, used when smoking cocaine, increases the risk of barotraumas such as pneumothorax, pneumomediastinum, and pneumopericardium.78,96,100 One study showed that 88% of patients presenting with pneumomediastinum had perceptible abnormal findings (such as subcutaneous emphysema or a Hamman's crunch) in their physical examination. Fifty-three percent of those cases were secondary to cocaine use.98 "Crack lung" refers to the triad of fever, bronchospasm, and pulmonary infiltrates.101Other signs that may be evident are acute pulmonary edema or heart failure. The latter may be either cardiogenic or due to pulmonary alveolar injury from impurities mixed with cocaine.102
All four extremities should be carefully evaluated for signs of limb ischemia. Case reports of peripheral arterial occlusion secondary to thrombosis have been reported with patients presenting, on average, 9.2 hours after using cocaine.103,104
A non-contrast head CT is indicated for patients with global or focal neurological symptoms not explained by routine bloodwork. In accordance with the American College of Emergency Physicians (ACEP) guidelines, a lumbar puncture (LP) should be performed in patients complaining of headache when the initial head CT is normal when considering subarachnoid hemorrhage (SAH).105,106 MRI/MRA should be considered in patients with persistent neurological complaints or deficits to rule out ischemia.
Given the high incidence of intracranial pathology, a non-contrast CT should be considered for patients presenting with a cocaine-associated seizure.107
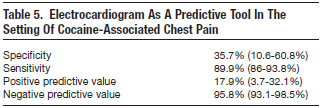
Patients presenting with cocaine-associated chest pain should have an ECG. This population may have a normal ECG 16-44% of the time. Conversely, ECGs yield abnormal or nonspecific results in 56-84% of these patients. 108-110 Many of these abnormal ECGs are manifestations of early repolarization and may be normal variations with J point and ST elevations. These findings occur in the septal leads greater than 85% of the time and rarely occur in limb leads.108,111 In fact, one study reported that 43% of patients with cocaine-associated chest pain had precordial ST-segment elevations meeting ECG criteria for use of thrombolytic therapy if ECG criteria were used alone.108 The sensitivity and specificity of ECGs in cocaine-associated chest pain for revealing acute MI are 36% and 90%, respectively5 (Table 5). Therefore, the ECG alone is an inadequate tool for diagnosing acute coronary syndrome in cocaine associated-chest pain. However, the ECG may be helpful if it demonstrates a dysrhythmia, differs from an old ECG, or evolves compared to the initial ECG.
It is important to note that ECG changes may persist long after cocaine use. Chakko et al found that 39% of ECGs were abnormal in 200 asymptomatic chronic cocaine users 72 hours after they were admitted to drug dependence treatment programs.112 ECG abnormalities were detected for two weeks in 25% of patients admitted for cocaine detoxification; this persisted for up to six weeks in some patients.68 Given the prevalence of the normal variants in this population, it is difficult to use the ECG as a precise tool in the evaluation of cocaine-associated chest pain.
The use of biochemical markers such as creatine kinase (CK), CK-MB, and cardiac troponin I (cTnI) have been used as an adjunct to the ECG for excluding MI in cocaine-intoxicated patients complaining of chest pain. CK and CK-MB have cross-reactivity with skeletal muscle and may be markedly elevated in the setting of cocaine use secondary to increased motor activity, trauma, rhabdomyolysis, and hyperthermia.
Most studies have shown cTnI to be more specific (94-100%) than CK-MB (75-88%) for identifying or excluding MI in the setting of cocaine-associated chest pain versus controls.113-115 A recent large prospective study found no statistical difference in the sensitivities (57% vs. 52%) and specificities (94% vs. 95%) of cTnI versus CK-MB.116 These results reinforce the recommendation to use cTnI to specify for myocardial necrosis as the marker of choice in patients with cocaine-associated chest pain. In this study, cTnI also had a good prognostic capability. Ninety-two percent of patients who had cTnI elevations had either a CKMB MI, significant disease on coronary angiography, or cardiac death. Thus, there is utility in pairing both these assays with a CK in cocaine-associated chest pain evaluations and little to no benefit in excluding a cTnI.
Myocardial perfusion imaging is commonplace for the evaluation of ischemia in patients who present with chest pain from traditional risk factors, and it has been proposed as a method for evaluating patients with chest pain in the setting of recent cocaine use. A prospective study, designed to evaluate resting myocardial perfusion using technetium-99m tetrofosmin in patients presenting with cocaine-associated chest pain with non-ischemic ECGs, found a low incidence (14%) of reversible ischemia.117 In a different study, only 5 of 216 patients presenting with cocaine-associated chest pain with low to moderate risk (defined as having non ischemic ECGs) had a positive result when evaluated by resting technetium-99m sestamibi (Tc-sestamibi) perfusion imaging. Two of the five patients with positive tests ruled in for MI. There were no cardiac events for study patients in a 30-day period following discharge.118 Although these data suggest that these imaging modalities are sensitive for myocardial perfusion defects, it remains to be seen whether or not this is a practical and cost-effective method of evaluating all low risk patients with cocaine-associated chest pain.
In summary, evaluations of patients that present with cocaine-associated chest pain should include serial ECGs, CK-MB, and cTnI as evidence of myocardial ischemia. Because of the low incidence of reversible ischemia in patients with cocaine-related chest pain, myocardial perfusion imaging should be reserved for those patients with intermediate or high risk factors.
A chest radiograph is indicated in all patients with cocaine-associated emergencies, especially in those with respiratory complaints. In addition to the fever and bronchospasm found on physical examination, "crack lung" typically includes evidence of diffuse alveolar infiltrates on chest radiography. Serum eosinophilia and elevated IgE levels suggest an immunological origin to these symptoms.119-121 Chest radiography may also identify cardiogenic and non cardiogenic acute pulmonary edema, pneumothorax, pneumomediastinum, hemothorax, pulmonary hemorrhage, or foreign body. In patients where thermal airway injury is suspected, a soft tissue neck radiography may reveal signs similar to infectious epiglottitis.90 In more severe cases, direct laryngoscopy/bronchoscopy may be indicated. CT of the chest and abdomen may be indicated if there is a high clinical suspicion of aortic dissection, occult pneumothorax, or gastrointestinal ischemia.
Patients presenting with psychomotor agitation should be checked for hypoxia, hypoglycemia, and hyperthermia. The latter is associated with a poor prognosis in animal models and should be managed with aggressive cooling measures such as ice packs and fanning. Violent activity should be immediately controlled with physical restraints until the patient can be chemically restrained. The prompt use of benzodiazepines decreases mortality in animal models, and they are the pharmacologic agents of choice for the treatment of cocaine-induced agitation.122,123 Dopamine antagonists, such as haloperidol, are contraindicated as they impair heat dissipation and increase mortality in experimental animal models.82,124 Because dantrolene does not act on the CNS, it does not ameliorate cocaine-induced hyperthermia.49,125,126
In addition to ameliorating the central manifestations of cocaine intoxication, benzodiazepines decrease the centrally-mediated and peripheral sympathomimetic outflow that contributes to the symptoms of cocaine-associated chest pain. Cocaine's ability to increase heart rate and systemic arterial blood pressure augments myocardial oxygen demand and consumption, primarily in the setting of coronary artery vasoconstriction. This exacerbation of the myocardial oxygen supply and demand mismatch was well depicted in a human cardiac catheterization study.127 Benzodiazepines decrease the myocardial workload by controlling the psychomotor hyperactivity, lowering the systemic arterial blood pressure, and reducing the heart rate.128
Benzodiazepine's effective treatment of both the central and peripheral manifestations of cocaine intoxication and their few side effects make them an excellent first-line therapy in patients with cocaine associated complaints.
The use of aspirin or other anti-platelet agents in the setting of cocaine-induced cardiac ischemia has not been studied. Aspirin acts to inhibit platelet aggregation, theoretically counteracting the pro-thrombotic properties of cocaine. Given the general safety record of aspirin use in patients with chest pain secondary to coronary artery disease, aspirin should be given to patients with cocaine-associated chest pain. It should only be withheld if there is a suspicion of intracranial hemorrhage or aortic dissection.
Nitroglycerin is a standard treatment for myocardial ischemia and decreases myocardial workload by lowering mean arterial pressure and maintaining myocardial perfusion. Cocaine-induced vasoconstriction is thought to be mediated through an α-adrenergic mechanism that nitroglycerin counteracts by acting directly on smooth muscle.129 Studies suggest that nitroglycerin plays a beneficial role in the setting of cocaine-associated chest pain. Brogan et al demonstrated angiographically a reversal of cocaine-induced coronary artery vasoconstriction with the administration of sublingual SL NTG in both diseased and nondiseased coronaries.130 A multi-center prospective observational study found nitroglycerin to be a safe and effective treatment of cocaine-associated chest pain.129There have been only two prospective randomized controlled trials comparing benzodiazepines, nitroglycerin, or both for the treatment of patients with cocaine-associated chest pain. 131,132 Only one of these studies found that the combination of nitroglycerin with lorazepam may be more efficacious than nitroglycerin alone.131 This study lacked a placebo group, potentially compromising both patients' and practitioners' objectivity.
α-adrenergic antagonists (such as phentolamine) have been demonstrated to reverse cocaine-induced vasoconstriction in experimental cardiac catheterization trials.127 Conversely, cocaine-induced vasoconstriction is potentiated by β-adrenergic antagonist agents; consequently, they are contraindicated in the treatment of cocaine-associated ischemia. Labetalol, a mixed α- and β-adrenergic blocker, reduces mean arterial pressure but does not ameliorate cocaine-induced coronary arterial vasoconstriction.133 In an animal study, labetalol administered to cocaine-poisoned rats was associated with higher rates of seizure and death.134Clinical trials demonstrate that β-adrenergic antagonist agents exacerbate unopposed cocaine-induced α-adrenergic stimulation and fail to effectively lower arterial pressure.135
Calcium channel blocking agents have an undefined role in the treatment of cocaine-associated chest pain. It is postulated that calcium channel blockers may attenuate the vasoconstrictive effects of cocaine. However, animal studies have yielded conflicting results. Rats pretreated with diltiazem, nifedipine, or verapamil before administration of cocaine developed seizures more rapidly than controls.136 Alternatively, nitrendipine had protective CNS effects in cocaine-fed rats.137A human clinical trial demonstrated that verapamil successfully reversed cocaine-induced coronary artery vasoconstriction and elevation in arterial pressure.138 Though this human data is encouraging, the study was limited to one calcium channel blocker and low doses of cocaine.
Much of the literature cautions against the routine use of thrombolytic therapy for patients with cocaine-associated MI. The selection of patients who meet the TIMI (thrombolysis in myocardial infarction) electrocardiographic criteria for thrombolytic therapy is hampered by the high rate of abnormal or nondiagnostic ECGs in patients presenting with cocaine-associated chest pain.108,111,139 Although a retrospective cross sectional survey of 25 patients found no major complications or deaths in patients with cocaine-associated MI who received thrombolytic therapy (95% CI 0% to 12%), fatal intracerebral hemorrhages after thrombolytic therapy in this setting does occur.140Due to the small number of patients and the lack of fatalities among the treated and the non treated patients, this study failed to demonstrate a benefit on survival. Similarly, an effect of thrombolytic therapy on infarct size could not be demonstrated by the insufficient CPK data. Those patients manifesting evidence of infarction that fail to respond to medical therapy (oxygen, aspirin, nitrates, benzodiazepines, phentolamine) should be considered for angioplasty. Thrombolysis may be used only when invasive therapy is unavailable and if there are no contraindications.141,142
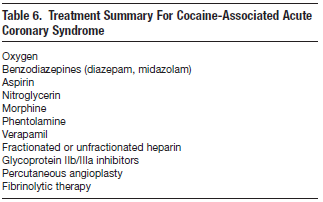
In summary, the medical treatment of presumed cocaine-associated myocardial ischemia should begin with the sequential treatment of oxygen, benzodiazepines, nitroglycerin, and aspirin as the diagnostic investigation is underway. The American Heart Association guidelines recommend nitroglycerin (Class I) and benzodiazepines (Class IIA) as first-line agents for cocaine-associated MI. Phentolamine (Class IIB) is considered a second-line agent for patients that do not respond to benzodiazepines and nitrates. Labetalol, a mixed α/β blocker, is a third-line agent, whereas nonselective β-blockers such as propranolol (Class III) are contraindicated.143 Patients who demonstrate myocardial ischemia by ECG or serum markers or who have hemodynamic instability should be considered for cardiac catheterization or thrombolysis when catherization is not available. They should also be admitted to a cardiac care unit.
The use of fractionated or unfractionated heparin and glycoprotein IIb/IIIa inhibitors in the setting of cocaine-induced myocardial ischemia has not yet been studied. They may be cautiously considered in patients demonstrating true myocardial ischemia once risks (such as intracerebral hemorrhage, trauma, and aortic dissection) have been excluded.
Cocaine induces cardiac dysrhythmias such as sinus tachycardia, wide complex supraventricular tachycardia, atrial fibrillation, and ventricular tachycardia. Cocaine propagates these disturbances by increasing catecholamine-induced ventricular irritability, action potential prolongation, and increased afterdepolarizations secondary to intracellular calcium concentrations.142 Cocaine blocks sodium channels and prolongs the QRS complex in a manner similar to Class IA antidysrhythmic agents and tricyclic antidepressants. In the setting of seizures and acidosis, sodium bicarbonate narrows cocaine-induced wide complex dysrhythmias and corrects the underlying acidosis. Sodium bicarbonate narrowed the QRS complex and corrected the acidosis in two case series of six patients presenting with cocaine-induced wide complex dysrhythmias. 69,70 Several animal studies have concurred with these findings. Therefore, sodium bicarbonate may correct the effect of cocaine on the sodium channel and should be considered in the management of cocaine-induced wide complex dysrhythmias. (Class IIA) Treatment with Class IA antidysrhythmic agents such as procainamide and quinidine may exacerbate prolongation of the QRS complex and
should be avoided.142
Lidocaine has many similarities to cocaine; both are sodium channel blockers and pro-convulsant anesthetics. Unlike cocaine, lidocaine is a Class IB antidysrhythmic agent and therefore exhibits faster on/off kinetics for binding to the sodium channel. This property may provide lidocaine an advantage as treatment for ventricular dysrhythmias that develop rapidly after cocaine use. However, the administration of lidocaine for cocaine-induced conduction disturbances has yielded conflicting results in animal models.145 There is only one human clinical study reporting the efficacy of lidocaine in cocaine-induced ventricular dysrhythmias.67In this retrospective case series of 29 patients who received lidocaine during cocaine-associated MI with ventricular tachycardia, no adverse CNS or cardiovascular outcomes were reported (95% CI 0% to 11%).144 Thus, lidocaine may be considered a Class IIB recommendation.
Acute gastrointestinal (GI) complaints associated with cocaine are uncommon and generally occur within the first 48 hours following cocaine use. Cocaine can affect the entire length of the GI tract causing dysphagia/odynophagia, nausea/vomiting to abdominal pain, and bloody diarrhea. These complications are associated with a 21% mortality rate.146 Chronic cocaine-induced atherosclerosis may cause symptoms of mesenteric ischemia such as postprandial abdominal pain and weight loss.147-149
α-adrenergic vasoconstriction of gastric and mesenteric arteries occurs after cocaine use. This decrease in mesenteric blood flow may result in bowel edema, ulceration, and necrosis. Perforation of the duodenum, jejunum, ileum, and colon has been described 146,150-153 as well as intra-peritoneal hemorrhage and splenic infarction.154-158
Though gastrointestinal ischemia occurs with all routes of cocaine administration, smoked cocaine is associated with gastroduodenal ulceration and perforation.146,147Appropriate imaging and surgical consultation should be obtained when bowel ischemia or perforation is suspected.
Body packers intentionally ingest cocaine in sealed packets for illegal transport. Body stuffers haphazardly swallow poorly packaged cocaine to conceal evidence. Both may present with acute gastrointestinal complaints and a variable severity of constitutional signs of cocaine poisoning.159 Plain radiographs detect foreign bodies in only 75% of body packers and are even less effective for body stuffers.160 Experts recommend observing asymptomatic patients in the ED. Symptomatic patients should be treated for acute symptoms with benzodiazepines and taken to the operating room for definitive extraction of drug packets. Asymptomatic body packers should be decontaminated (activated charcoal 1 g/kg body weight), whole bowel irrigated (electrolytes/polyethylene glycol 1.5-2 L/hr), and admitted to the hospital. CT scan should be used to confirm full extraction of drug packets, and surgical consult should be on standby in case the patient becomes symptomatic.161
Cocaine can precipitate acute renal failure through a variety of mechanisms. Catecholamines potentiate malignant hypertension, vascular thrombogenicity, and vasospasm in the renal vasculature. These mechanisms have been implicated in cases of cocaineassociated renal failure without rhabdomyolysis.162 Cases of renal infarction have been reported after cocaine use, though they are rare.163,164
Cocaine is associated with traumatic and atraumatic rhabdomyolysis and acute tubular necrosis leading to renal failure.165 A prospective study of patients presenting to the ED with cocaine-related complaints found that only 13% experienced the classic signs of rhabdomyolysis (nausea, vomiting, muscle pain/weakness/tenderness), with 24% having elevated CK levels greater than 1000 U/L.166 Patients with normal creatinine levels, normal WBC, and CK levels less than 1000 U/L had a decreased incidence of renal complications.167
Treatment should be aimed at attenuating the centrally mediated excitatory state with benzodiazepines, aggressive blood pressure control, and IV hydration. Rhabdomyolysis may require saline, mannitol administration, and urinary alkanization. Treatment of cocaine-associated acute renal failure with dopamine and furosemide was found to hasten recovery in one study.168
Cocaine crosses the placenta and results in fetal growth retardation, neurological abnormalities, genitourinary abnormalities, prematurity, smaller birth rate, and increased risk of sudden infant death syndrome (SIDS). 169-172
Cocaine use by gravid women complicates pregnancy and negatively affects post-partum development of the child. Hypertension, increased uterine vascular resistance, and increased uterine contractility decreases uterine blood flow.173,174 This may lead to spontaneous abortion, placental abruption, premature labor, uterine rupture, and conditions that resemble preeclampsia.175-177 One study found that 24% of the risk of spontaneous abortion is attributable to cocaine and tobacco; however, since many women may mistake a pregnancy with a missed menses, the true percentage may remain elusive.178
Chronic cocaine insufflation leads to local necrosis and nasal septum perforation. More extreme cases have extended to involve oronasal fistulas, orbital cellulitides, nasolacrimal duct obstruction, and skull base destruction with pituitary infarction.179-182
Ocular complaints in patients with chronic or recent cocaine abuse should prompt a slit lamp examination and measurement of intraocular pressure to rule out cocaine-associated acute angle glaucoma and ulcerative keratitis. 183,184 Fora complete description of these procedures, subscribers can click here to view our September 2007, Volume 9 Number 9 issue, An Evidence-Based Approach To Abnormal Vision.
Early research on patients presenting with cocaine-associated chest pain established the hazards of its use. However, several questions remained unanswered. Which of these patients will need admission? Which of these patients may be safely discharged? How long is a safe observation period for those patients in the ED that will be discharged? What kind of tests should they have during this observation period? These questions have been the subject of recent research.
Patients presenting with cocaine-associated chest pain have approximately a 6% incidence of MI.185 In a study of 130 patients, there was a 0% hospital mortality (95% CI 0% to 3%).186 Furthermore, a retrospective study found that only 36% of patients with cocaine-associated MI diagnosed by serum markers have complications (defined as congestive heart failure, ventricular tachycardia, supraventricular tachycardia, and bradydysrhythmias). Forty-eight percent of these complications are present on arrival to the ED, and 90% are evident within the first 12 hours of presentation.180
A previous study had already calculated the one-year actuarial survival as 98%, suggesting that low to intermediate risk patients with cocaine-associated chest pain were unlikely to benefit from hospital admission. A retrospective study of patients with cocaine-associated chest pain who were studied with Tc-sestamibi supported these figures. In this study, low risk patients who underwent testing were not identified as having any cardiovascular complications after a 30-day follow-up period.107
Prospectively, a case series demonstrated the safety of dobutamine stress chocardiography for low-risk patients presenting to the ED within 24 hours of cocaine use. Pharmacological stress testing has been proposed to help assess and stratify patients with cocaine-associated chest pain; however, there has been concern about evoking an exaggerated adrenergic response. In this study, most subjects reached their target heart rates and none of the subjects exhibited an exaggerated adrenergic response or developed tachydysrhythmias.187
The safety of this approach was further supported by another prospective study by Weber et al. In this study, 302 patients presenting with cocaine-associated chest pain were stratified into low to intermediate and high-risk groups. High-risk patients, defined as those whose initial ECG suggested ischemia or MI, an ECG with 1 mm or more ST segment elevation or depression (or elevated serum cardiac markers), recurrent ischemic chest pain, or hemodynamic instability were directly admitted. Intermediate to low risk patients underwent continuous 12 lead cardiac monitoring and serum cTnI evaluations at three, six, and nine hours. Those patients without recurrent symptoms or evidence of myocardial necrosis or ischemia after nine hours of observation underwent an exercise stress test according to the Bruce protocol. Because very few of these patients had positive tests, the protocol was changed to recommend outpatient stress testing. There were no cardiovascular deaths during the observation period or during the 30-day follow-up period. Non fatal MI only occurred in patients with continued cocaine use (1.6%). The outcomes in this study are similar to those reported for 9-12 hour protocols in non-cocaine using patients with traditional risk factors.188
Some investigators have noted that there may be an underlying high incidence of false positive stress tests in this population. They recommend that there be a 12-day delay after the cessation of cocaine use for the stress-testing.189
In all these studies, death or non fatal MI occurred only in patients with continued cocaine usage or in patients who had preexisting conditions.188,190 This data suggests that low to intermediate risk patients with cocaine-associated chest pain are unlikely to benefit from hospital admission and that, in addition to medical follow-up, cocaine cessation is an essential part of their post-discharge planning.
There are no data or guideline recommendations for the disposition of cocaine-associated neurological complaints presenting to the ED to distinguish them from non cocaine-associated neurological complaints. These patients should be treated with a high degree of clinical suspicion and should have aggressive work up with definitive imaging. Patients with persistent neurological symptoms or deficits should have a neurology consult if available and should be admitted.
Since many patients with cocaine toxicity will present with chest pain, it is essential that these patients be recognized early and managed appropriately, using risk stratification. Low to intermediate risk patients represent the majority of patients presenting with cocaine-associated chest pain. They can be safely discharged from the emergency department at the end of a 9- to 12-hour observation period and after obtaining two normal serial serum cTnI levels and non ischemic ECGs. For high-risk patients, a cardiology consultation and admission to the cardiac care unit should be considered.
The prevalence of cocaine use in the United States remains high despite efforts to curb its availability. Consequently, there are an increasing number of patients presenting to EDs with a variety of cocaine-associated complaints, posing a challenge to emergency physicians.
Neurological complaints should be evaluated aggressively. Low risk patients that have symptom resolution and negative imaging may be discharged, while those with persistent symptoms or imaging abnormalities warrant a neurology consult and hospital admission. Chest pain patients should be stratified into high-, medium-, and low-risk groups. High-risk patients with cocaine-associated chest pain should be admitted to a coronary care unit, especially if they demonstrate evidence of acute MI, dysrhythmia, or hemodynamic instability. Low- to intermediate-risk patients may be discharged from the ED or observation unit after a 9- to 12-hour observation period utilizing the biomarker, cTnI, and ECG analyses. These patients are at very low risk for immediate complications and may receive further cardiac evaluation in the outpatient setting. The ED visit is an important time to commence cocaine cessation initiatives.
The patient in police custody became belligerent when he was refused smoking and telephone privileges. Multiple doses of lorazepam were required to achieve sedation and normalization of his vital signs. Ethanol and creatine kinase were elevated. Troponin was negative and the chest x-ray was normal. He was hydrated with 3 liters of normal saline and received aspirin. After 12 hours of observation, the creatine kinase normalized, the second troponin was negative, and the lateral T wave inversions on the initial ECG were unchanged. The patient denied any complaints on reevaluation and was now politely asking for a smoke. He was discharged with a referral for detox. The second patient, Fred, had a second generalized tonic-clonic seizure as the nurse was attempting intravenous access. He was given intramuscular midazolam which terminated the event. An emergent CT of the head revealed a small SAH in the left parietal lobe without mass effect or shift. The patient was accepted for transfer to the University Medical Center. When you entered the waiting room, the cell-phone chatter paused. Fellow students looked on, mouths agape, as Fred was loaded into the transport ambulance.

 |
|
|
|
|
 |
 |
 |
Evidence-based medicine requires a critical appraisal of the literature based upon study methodology and number of subjects. Not all references are equally robust. The findings of a large, prospective, randomized, and blinded trial should carry more weight than a case report.
To help the reader judge the strength of each reference, pertinent information about the study, such as the type of study and the number of patients in the study, will be included in bold type following the reference, where available. In addition, the most informative references cited in this paper, as determined by the authors, will be noted by an asterisk (*) next to the number of the reference.
Peter K. Dittmar; Ruben Olmedo
January 1, 2008
Emergency Medicine Practice • CONTINUE READING
Access every issue, our complete clinical pathway library, and earn up to 190 CME credits with an annual subscription.
Stay current with a new Emergency Medicine Practice issue every month, plus unlimited access to our complete issue library, all Interactive Clinical Pathways, and up to 190 CME credits.
Accredited By





Our Partners


 678-366-7933
678-366-7933