
|
|
Most emergency clinicians are quite comfortable treating diabetic ketoacidosis (DKA) in children, but other rarer endocrine disorders in this population are likely to cause anxiety in even the most well-read emergency clinician. In addition to their complex pathophysiologies, these disorders present with an array of nonspecific complaints — the most ominous of which is an altered mental status. This issue of Pediatric Emergency Medicine Practice reviews the diagnosis and management of these uncommon disorders, which, if left untreated, can cause significant morbidity.
It’s 2:30 am on a slow Thursday night when the triage nurse brings in an ill-appearing, tachypneic, and febrile 2-year-old with markedly delayed capillary refill. As you are listening to inspiratory crackles in the child’s left lung, you notice that her medications list includes hydrocortisone and fludrocortisone. You ask the child’s mother about these uncommon medications, and she informs you that her daughter has congenital adrenal hyperplasia. As you struggle to recall the specifics of this relatively rare condition, the nurse asks you whether this diagnosis is going to change the management of this critically ill child.
ACTH: Adrenocorticotropic hormone
ADH: Antidiuretic hormone
BUN: Blood urea nitrogen
CAH: Congenital adrenal hyperplasia
CNS: Central nervous system
CSW: Cerebral salt wasting
DKA: Diabetic ketoacidosis
GI: Gastrointestinal
ED: Emergency department
EMS: Emergency Medical Services
FSH: Follicle-stimulating hormone
ICU: Intensive care unit
IM: Intramuscularly
IV: Intravenously
LH: Luteinizing hormone
MMI: Methimazole
PO: “per os” by mouth
PTO: Propylthiouracil
SIADH: Syndrome of inappropriate antidiuretic hormone
T3: Triiodothyronine
T4: Thyroxine
tid: “ter in die” three times a day
TRH: Thyrotropin-releasing
TSH: Thyroid-stimulating hormone
A literature search was performed using Ovid MEDLINE® and PubMed. Keywords included adrenal insufficiency, congenital adrenal hyperplasia, 21-hydroxylase deficiency, and stress-dose corticosteroids. Similar searches were performed in the Cochrane Database of Systematic Reviews and the National Guideline Clearinghouse.
Most of the literature on pediatric adrenal insufficiency published within the past 20 years has addressed its 2 main causes: congenital adrenal hyperplasia and adrenal suppression due to chronic corticosteroid use. Unfortunately, most of these articles are case series or observational studies, with few clinical trials. Within the last decade, adrenal insufficiency in critically ill children has become a hot topic, resulting in some well-designed studies that are covered in this article.
Acute adrenal insufficiency occurs when the adrenal cortex fails to produce enough cortisol in response to stress, which is often triggered by infection or trauma. Patients classically present with inappropriate and rapid decompensation in the presence of a stressor, but in some cases, symptoms develop with no obvious inciting event.1 The most common cause of acute adrenal insufficiency in North America is the sudden discontinuation of or noncompliance with medication or emesis in patients who are on long-term glucocorticoid therapy.2 Adrenal insufficiency also occurs in patients receiving such therapy who are subjected to stressors such as sepsis, trauma, or surgery. Although it is seen most commonly in patients who are taking long-term oral glucocorticoids, significant adrenal suppression has also been described with chronic use of high-dose inhaled, topical, and intranasal preparations.3-7
Congenital adrenal hyperplasia (CAH) is an inherited defect of cortisol synthesis and the most common cause of primary adrenal insufficiency in children.1 A description of the pathophysiology of CAH is beyond the scope of this article, but it should be noted that these patients require long-term glucocorticoid replacement and many also require mineralocorticoid replacement. Newborn screening for CAH is now carried out in most parts of the United States.8 Many females with this disorder can be identified clinically at birth because of the associated virilization of their genitalia. In contrast, in unscreened males, the diagnosis is often not made until school age or later.9-11
The pertinent anatomy and physiology of the adrenal gland are shown in Figures 1 and 2.
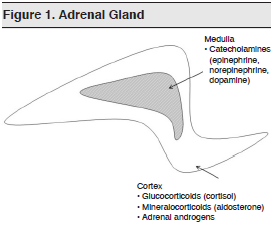
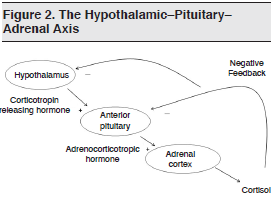
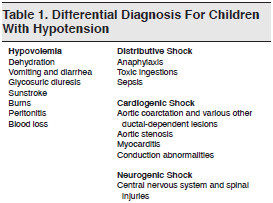
Children with acute adrenal insufficiency classically present with dehydration, hypotension, hypoglycemia, or altered mental status.1 Since these episodes are often triggered by infection or trauma — conditions which by themselves may produce these nonspecific findings — making the diagnosis of acute adrenal insufficiency is extremely difficult on the basis of clinical signs alone. Consider adrenal insufficiency when reviewing the differential diagnoses for children with hypotension and altered mental status. (See Tables 1 and 2.)

Given its rarity and nonspecific symptoms, pediatric adrenal insufficiency is unlikely to be recognized by Emergency Medical Services (EMS) personnel. However, many children with acute adrenal insufficiency initially present with hypotension, hypoglycemia, and altered mental status — symptoms frequently encountered in the prehospital setting. Fluid resuscitation with isotonic saline, correction of hypoglycemia, and a standard protocol for altered mental status should be initiated.
Children with acute adrenal insufficiency will frequently require resuscitative efforts in the emergency department (ED). One of the first clues to adrenal insufficiency may be hypotension unresponsive to appropriate fluid resuscitation, such as in a child with presumed sepsis. As with any child with hypotension due to dehydration, 20 mL/kg of isotonic saline boluses should be administered intravenously (IV) until adequate tissue perfusion is restored. Hypoglycemia is common in young children1 and can be corrected by infusing 5 mL/kg of dextrose 10% in water (D10) in infants, 2 mL/kg of D25 in toddlers, and 1 mL/kg of D50 in older children. Of note, children with presumed sepsis who require intubation should not receive etomidate as an induction agent. Etomidate-induced adrenal suppression is a welldescribed phenomenon in adults, and 2 pediatric studies have documented an increased risk of death in children with sepsis after a single bolus of etomidate during intubation.24,83
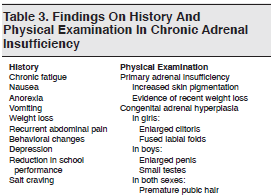
It is vital to determine whether there is a history of corticosteroid use when confronted with a child who is clinically unstable or appears to have sepsis. Although it is difficult to reliably estimate the degree of adrenal suppression based on the dose or duration of corticosteroid therapy, a treatment course longer than 2 weeks is typically necessary to suppress endogenous cortisol production.1,25 Adrenal suppression may persist for 6 to 9 months after long-term glucocorticoid therapy has been discontinued. Patients taking corticosteroids for CAH or other conditions may be wearing an identification bracelet indicating their condition. Common symptoms of chronic adrenal insufficiency are listed in Table 3.1,26-28
The physical findings in acute adrenal insufficiency are typically characteristic of the precipitating illness rather than specifically suggestive of adrenal insufficiency. Hypotension unresponsive to fluid resuscitation is characteristic of, although not pathognomonic for, acute pediatric adrenal insufficiency.22,23 Other findings include an altered mental status, orthostatic hypotension, and other signs of dehydration such as sunken eyes and dry mucous membranes.
The physical findings in chronic adrenal insufficiency vary widely depending on the cause. In children with primary adrenal insufficiency, increased skin pigmentation is a key finding; it is most pronounced in scars, flexion creases, areolae, gums, and nonexposed areas of skin. Physical findings in primary adrenal insufficiency and CAH are listed in Table 3.1,27,28 Children with adrenal suppression from chronic corticosteroid use may have cushingoid features (eg, truncal obesity, rounded faces, striae, and acne).
A chemistry panel may be the most useful routine test when considering the possibility of childhood adrenal insufficiency. Hypoglycemia is the most consistent laboratory finding in young children.1 Low serum sodium levels (hyponatremia) and high serum potassium levels (hyperkalemia) will typically be present in cases of primary adrenal failure or CAH when the production of both cortisol and aldosterone is impaired. Other classic findings of aldosterone insufficiency include a reduced serum bicarbonate level and an increased chloride level (a non anion-gap metabolic acidosis). Secondary adrenal failure due to withdrawal from chronic glucocorticoid therapy or to adrenocorticotropic hormone (ACTH) deficiency will present with cortisol deficiency only.
Ideally, adrenal insufficiency is a diagnosis to be suspected in the ED and confirmed after admission to the hospital. Once suspected, it is important to collect urine and blood for confirmatory testing before administering glucocorticoids, if at all possible,1,27,28 and it is wise to consult with a pediatric endocrinologist when ordering these highly specialized tests. (See Table 4.) At some institutions, serum cortisol can be measured in the ED. A random cortisol value may not be helpful in ruling out adrenal insufficiency, since a single low value does not definitively confirm the diagnosis; however, a serum cortisol concentration less than 10 mcg/dL in children who are not acutely ill or less than 18 mcg/dL in those who are acutely ill is highly suggestive of adrenal insufficiency.1,28,33-35,82

The first priority in children with presumed adrenal insufficiency is restoration of tissue perfusion and correction of hypoglycemia, as previously discussed. Some patients with primary adrenal insufficiency will have clinically significant hyponatremia and hyperkalemia due to aldosterone deficits. Patients with a serum potassium level greater than 6 mEq/L should be treated with one or several of the options listed in Table 5.27

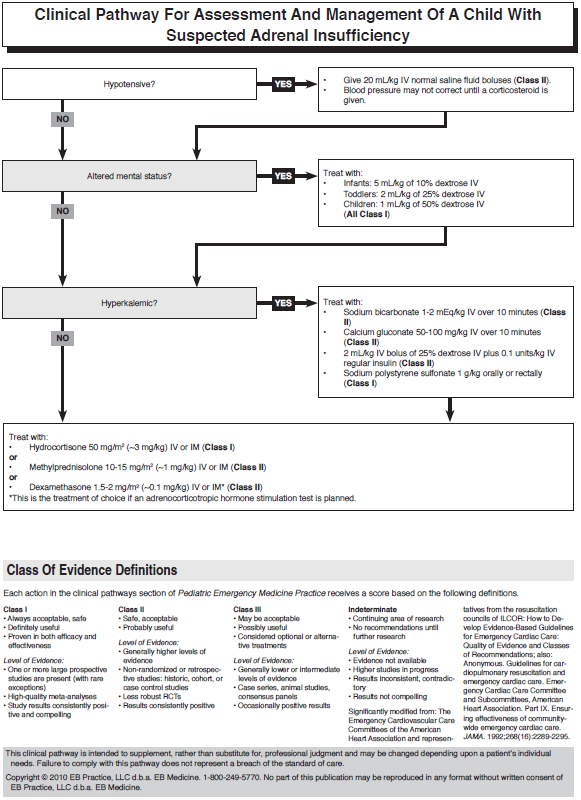
Very few pediatric studies have looked at corticosteroid dosing in children with acute adrenal insufficiency, and most current recommendations are empiric and extrapolated from adult studies. Hydrocortisone at a dose of 50 mg/m2 (about 3 mg/kg) IV or intramuscularly (IM) is recommended by several authors as the initial treatment of choice.1,27,28,37,82 Hydrocortisone has a mineralocorticoid effect in addition to its glucocorticoid effect (eg, 30 mg of hydrocortisone has the mineralocorticoid effect of 0.1 mg of fludrocortisone). Methylprednisolone 10 to 15 mg/m2 (about 1 mg/kg) IV or IM or dexamethasone 1.5 to 2 mg/m2 (about 0.1 mg/kg) IV or IM may also be given, although neither of these agents has intrinsic mineralocorticoid activity. If the child can tolerate oral medication, fludrocortisone (0.1-0.2 mg) can be given to provide mineralocorticoid activity if primary adrenal failure is suspected.
Some controversy exists as to when and how to give “stress doses” of corticosteroids to patients on chronic corticosteroid therapy. Current recommendations are to double or triple the daily dose in patients with simple febrile illnesses, such as an upper respiratory infection or streptococcal pharyngitis, for the duration of the illness.38-40 Children who cannot be given oral medication because of vomiting or those being treated just prior to surgery can be given IV hydrocortisone as follows: 25 mg for children under 3 years of age, 50 mg for children ages 3 to 12, and 100 mg for adolescents. Following the initial dose, these children should receive the same amount per day in divided doses. Children with more severe and life-threatening conditions, such as pneumonia, meningitis, or major trauma, require higher doses of IV hydrocortisone, up to 100 mg/m2/day (about 7 mg/ kg) divided every 6 hours.10,38,39,41
With rare exception, children with new-onset adrenal insufficiency will require admission to the hospital. Often the disease process that is exacerbating the insufficiency will itself warrant admission (eg, gastroenteritis with dehydration or pneumonia with hypoxia). Children who are hemodynamically unstable (ie, a serum sodium level less than 125 mg/dL or a serum potassium level greater than 6 mEq/dL) must be admitted to an intensive care unit (ICU).
Children with known adrenal insufficiency who present with a mild illness (eg, otitis media) and no clinical evidence of acute adrenal insufficiency can be managed as outpatients, as previously discussed, if they are able to tolerate oral medication without emesis. Close follow-up, preferably within 48 hours, should be arranged for these children prior to ED discharge. Instruct parents to return if the child’s condition worsens, specifically in the case of altered mental status or excessive vomiting.
A literature search was performed using Ovid MEDLINE ® and PubMed using the keyword pheochromocytoma. Similar searches were performed of the Cochrane Database of Systematic Reviews and the National Guideline Clearinghouse. The vast majority of papers published on pediatric pheochromocytoma in the past 40 years consists of case series. Given the rarity of this illness, clinical trials focused on its management would be extremely difficult.
Pheochromocytomas are rare childhood tumors that arise from the chromaffin tissue of the adrenal medulla and sympathetic ganglia.12 Pheochromocytomas originating outside the adrenal gland are often called paragangliomas. Pheochromocytomas are malignant in 12% to 40% of childhood cases, and the average age at presentation is 9 to 11 years.13-17 Pediatric pheochromocytomas are associated with familial syndromes, including multiple endocrine neoplasia, Sipple’s syndrome, Sturge–Weber syndrome, von Hippel–Lindau disease, tuberous sclerosis, and neurofibromatosis.13,14,16-21 Excess catecholamine secretion from pheochromocytomas causes symptoms such as hypertension, tachycardia, and headaches.
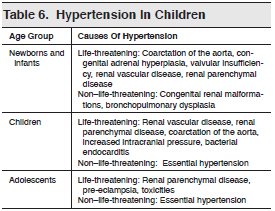
Children with pheochromocytoma most commonly present with signs and symptoms related to hypertension.15 The presence of hypertension in a child with otherwise nonspecific complaints opens up a broad differential diagnosis. (See Table 6.)
The most common presenting symptoms related to catecholamine excess in children include headache, usually described as throbbing, and inappropriate sweating.12,13,16,17,21,29-31 Other symptoms of pheochromocytoma are listed in Table 7. Consider pheochromocytoma when presented with children who have these nonspecific symptoms and a known multiple endocrinopathy or neurocutaneous syndrome.
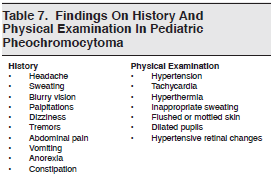
Sustained hypertension is the most consistent physical finding in children with pheochromocytoma, being present 88% to 93% of the time.12,13 Other physical findings are listed in Table 7.13,15-18,29,31 Of note, in a small percentage of cases of pediatric pheochromocytoma, the tumor mass will be palpable in the neck or abdomen.12,31,32
Normal results on a chemistry panel may help distinguish pediatric pheochromocytoma from renal causes of childhood hypertension, and fasting blood sugar will typically be elevated in children with this tumor.17
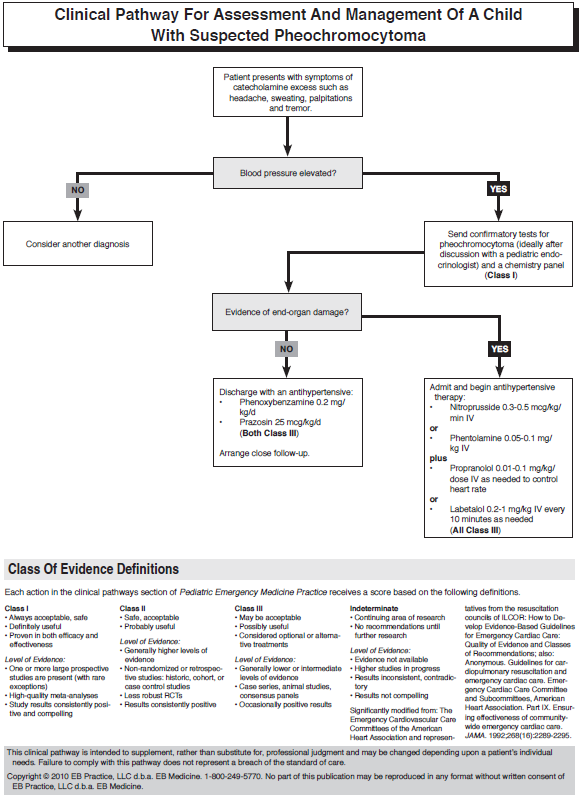
As with adrenal insufficiency, the emergency clinician’s role in diagnosing pediatric pheochromocytoma is primarily to consider it when the setting seems appropriate. Once suspected, diagnostic studies are of 2 types: biochemical and anatomic. The biochemical diagnosis relies on detecting an elevated level of catecholamines or their metabolites in the blood or urine and can establish the diagnosis in over 95% of patients.15 Since pheochromocytomas are a heterogeneous group of catecholamine-secreting tumors, no biochemical test is 100% sensitive.13 The confirmatory blood and urine tests for pheochromocytoma are listed in Table 8.36 The availability of these tests varies by institution, and it is wise to consult a pediatric endocrinologist before ordering them. Imaging studies to determine tumor location (ideally magnetic resonance imaging) are obtained at a later date if results of initial biochemical tests are positive.

Control of blood pressure in children with suspected pheochromocytoma is necessary to reduce the risk of end-organ damage until definitive treatment (ie, surgical resection) is possible. Unfortunately, no controlled studies in children have been published comparing the efficacy of antihypertensives used for this purpose. In children requiring admission, IV nitroprusside at a starting dose of 0.3 to 0.5 mcg/ kg/min or phentolamine at a dose of 0.05 to 0.1 mg/ kg has been used at some institutions, with the addition of a beta blocker such as propranolol 0.01 to 0.1 mg/kg/dose IV, if needed, to control the heart rate.17,20 Labetalol (0.2-1 mg/kg IV every 10 minutes as needed) has both alpha- and beta-blocking effects and has been used by some centers, although others have had little success with it.16,21,31,42 In children who are to be discharged pending further work-up, phenoxybenzamine at a starting dose of 0.2 mg/kg/ day is advocated in many sources.15-21,30,42,43 Calciumchannel blockers and prazosin at a starting dose of 25 mcg/kg/day are other outpatient options.15-17,31,43 pheIdeally, the choice of an antihypertensive agent for these children should be discussed with a pediatric endocrinologist or nephrologist.
Children with suspected pheochromocytoma require admission if there is any clinical or laboratory evidence of end-organ damage. Children with mild symptoms may be discharged if appropriate follow-up has been arranged, ideally with a pediatric endocrinologist. Outpatient antihypertensive therapy should be discussed with the referral physician and should be begun before the child is discharged from the ED.
A literature search was performed using Ovid MEDLINE® and PubMed. Keywords included diabetes insipidus, SIADH, cranial salt-wasting, hypernatremia, and hyponatremia. Articles and studies from the pediatric literature were selected preferentially. Similar searches were performed of the Cochrane Database of Systematic Reviews and the National Guideline Clearinghouse.
Pediatric syndrome of inappropriate antidiuretic hormone (SIADH) and diabetes insipidus have not been well studied. Most publications on these topics have been case series, with a few studies addressing long-term treatment, and many of the management recommendations discussed in review articles are extrapolated from the adult literature. Perhaps of greater importance to the emergency clinician are the results of some good animal and human research on the treatment of symptomatic hyponatremia and hypernatremia.
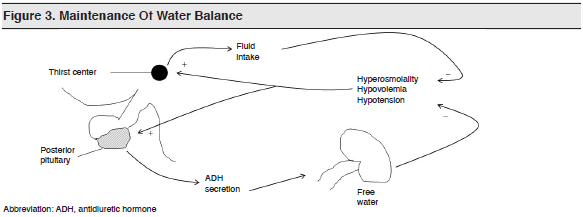
Antidiuretic hormone (ADH) is produced by neurosecretory neurons that originate in the hypothalamus and extend into the posterior pituitary gland. Water balance in the body is regulated by ADH and thirst. (See Figure 3.)
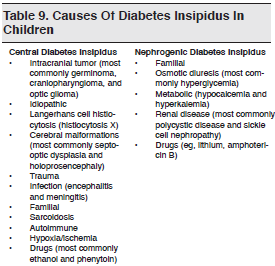
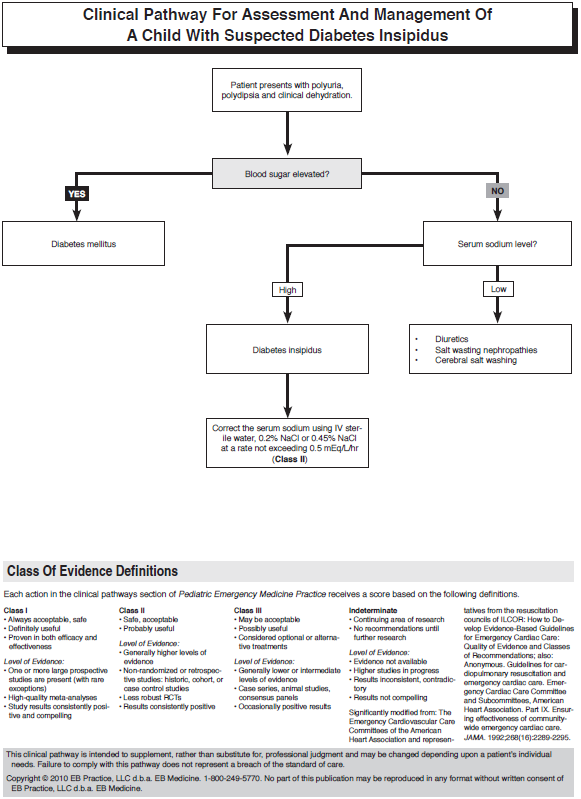
Diabetes insipidus is a condition defined as the passage of large volumes of dilute urine, in excess of 150 mL/kg/day in newborns, 110 mL/kg/day at 2 years of age, and 40 mL/kg/day in older children.44 Diabetes insipidus can be divided into 2 main categories: central diabetes insipidus, caused by inadequate secretion of ADH, and nephrogenic diabetes insipidus, characterized by the inability of the kidneys to concentrate urine in response to ADH. Not surprisingly, polydipsia, polyuria, and symptoms of dehydration are the main symptoms. Table 9 lists the causes of diabetes insipidus in children. Intracranial tumors and idiopathic causes have been the main etiologies of pediatric diabetes insipidus in most published case series.45-48 It has been observed that many children with central diabetes insipidus also have dysfunction of the anterior pituitary, with deficiencies of thyroid-stimulating hormone (TSH), growth hormone, follicle-stimulating hormone (FSH), and luteinizing hormone (LH) or ACTH.45,48-50
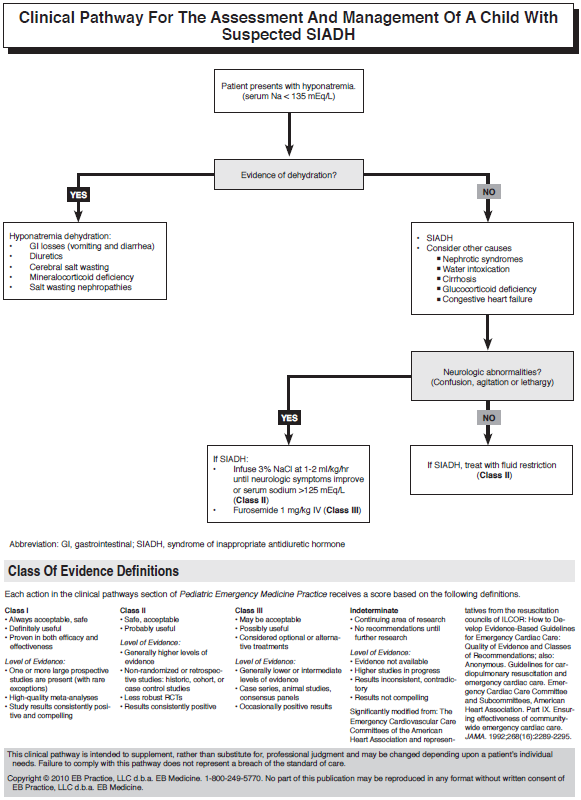
Syndrome of inappropriate antidiuretic hormone is caused by excessive secretion of ADH and is uncommon in children. Excess ADH results in increased free water reabsorption by the kidneys, with subsequent hypervolemia and dilutional hyponatremia. A “nephrogenic syndrome of inappropriate antidiuresis” has also been described in infants; the clinical picture is the same as that of SIADH but with low serum ADH levels.51 Clinical criteria used to diagnose SIADH are listed in Table 10.52 Syndrome of inappropriate antidiuretic hormone in children is usually a transient and self-limited phenomenon, with only a few case reports describing it as a chronic condition.53,54 The causes of SIADH in children are listed in Table 11.52,55 Although these causes have not been formally studied, some authors believe that the most common cause is the administration of vasopressin or its synthetic analogue, desmopressin.56 These medications are commonly used to treat diabetes insipidus, von Willebrand disease, hemophilia, and bed wetting.


Intense polyuria and polydipsia are the most common symptoms in children with diabetes insipidus. These symptoms may not be as readily apparent in infants who are more likely to present with poor growth and symptoms of dehydration.57 New-onset diabetes mellitus with uncontrolled hyperglycemia is a much more common cause of this clinical picture in children. Other conditions that present with symptoms similar to those of diabetes insipidus are listed in Table 12.

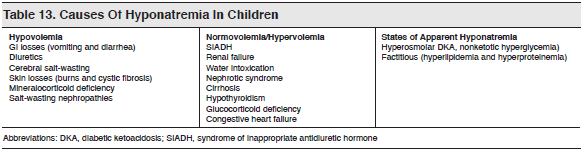
Symptoms and signs of hyponatremia without evidence of hypovolemia predominate in children with SIADH. Table 13 lists other conditions that can cause hyponatremia in children.58,59 The symptoms of hyponatremia in children are generally nonspecific and include anorexia, vomiting, lethargy, and weakness. More severe cases may present with obtundation and seizures. Syndrome of inappropriate antidiuretic hormone is an uncommon cause of hyponatremia in children, with gastrointestinal losses and water intoxication being much more common. Consider SIADH when treating a child with hyponatremia but no clinical signs of dehydration.
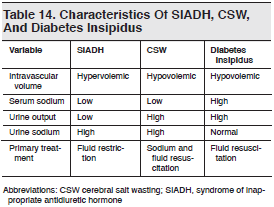
Cerebral salt-wasting (CSW) is a recently defined clinical entity with characteristics of both diabetes insipidus and SIADH.60 (See Table 14.) Like diabetes insipidus and SIADH, CSW can occur after central nervous system (CNS) insults such as trauma, infection, surgery, or tumors.61,84 Symptoms typically begin within 1 week of the insult and last for 2 to 4 weeks. Cerebral salt wasting is thought to be caused by atrial natriuretic peptide, a hormone which decreases the tubular reabsorption of sodium in the kidney, resulting in increased urine and sodium excretion. The net result is the inability to prevent sodium and water loss despite the presence of hyponatremia and hypovolemia. Cerebral salt wasting is marked by low serum sodium, high urine output, and renal salt loss. This is in contrast to SIADH in which hyponatremia is accompanied by low urine output. It is important to distinguish between SIADH and CSW, since the fluid management of each differs considerably.
Many children with diabetes insipidus will present with characteristic signs of dehydration. Treat this type of hypovolemia in standard fashion with 20 mL/kg isotonic saline boluses until adequate fluid volume has been restored.
The most critically ill children with SIADH will present with signs of cerebral edema and herniation caused by hyponatremia. Seizures and respiratory arrest are the most serious manifestations.59 Seizures should be initially managed in standard fashion with supplemental oxygen, rapid glucose testing, and the administration of benzodiazepines. (Keep in mind that the hyponatremic etiology of the seizure will rarely be apparent until after formal laboratory testing.) Respiratory arrest, even when managed in an appropriate fashion, has an exceptionally grim prognosis.59
Most older children with diabetes insipidus present with the classic symptoms of polyuria and polydipsia. Nocturia and new-onset bed wetting may also be present. Infants are more likely to present with symptoms of dehydration and poor growth, since they don’t have free access to fluids.57 Consider diabetes insipidus in children with these symptoms and a known intracranial tumor, cerebral malformation, or recent CNS infection or trauma. Ask about a family history of similar illnesses in children with these symptoms, since 5% of cases of diabetes insipidus in children are familial.45 In reality, diabetes insipidus will probably be the first diagnosis considered in the child with classic symptoms of new-onset diabetes mellitus who has a surprisingly normal blood glucose level.
Symptoms of hyponatremia predominate in children with SIADH. Most children remain asymptomatic until the serum sodium level drops below 125 mEq/L,55,62,63 and symptoms are more likely to become apparent when levels have dropped rapidly. Unfortunately, the symptoms of hyponatremia are netrather nonspecific. Headache, nausea, vomiting, and generalized weakness are the most consistent findings.55,58,59,62 Progressive neurologic abnormalities include lethargy, confusion, and agitation as hyponatremic encephalopathy progresses. Consider SIADH when a child is found to be hyponatremic in combination with a recent history of a CNS insult or use of desmopressin.
In children with diabetes insipidus, physical findings, if any, reflect a volume-contracted, hypernatremic state. Dry mucous membranes, sunken eyes, tachycardia, and listlessness may be present. In severe cases, the child may be comatose, with reduced skin turgor, causing the abdominal skin to have a doughy texture. Hypernatremia in children may result in increased muscle tone, nuchal rigidity, and brisk reflexes.
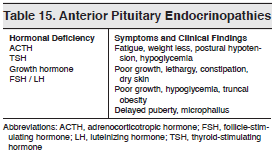
Infants with diabetes insipidus can have a failure-to-thrive appearance with wasted extremities and a protuberant abdomen. Vision may be impaired in children whose diabetes insipidus is caused by an intracranial tumor. Since many children with diabetes insipidus have associated anterior pituitary dysfunction, look for signs of coexisting endocrinopathies. (See Table 15.)
The physical findings in SIADH are primarily due to hyponatremia, with most signs indicative of CNS dysfunction. Generalized weakness, hyporeflexia, dilated pupils, and progressive alteration of mental status from mild confusion to coma typically occur.59,63 Note that these symptoms occur in children without evidence of dehydration, a fact that distinguishes SIADH from many other causes of hyponatremia in children.
Hypernatremia is typically present in infants with diabetes insipidus owing to the loss of free water. In older children, who are able to increase their fluid intake, serum sodium may not be elevated. Children who are dehydrated on presentation will usually have an elevated blood urea nitrogen (BUN)-to-creatinine ratio (> 20:1), as is typical of a volume-depleted state.64 As previously stated, a normal serum glucose quickly rules out diabetes mellitus.
The chemistry panel in children with SIADH reflects their diluted, hypervolemic state. Hyponatremia (serum sodium < 135 mEq/L) is present by definition. The BUN-to-creatinine ratio is usually less than 10:1.54,65
Although this test is neither sensitive nor specific for either of these disorders, a urine specific gravity can usually be measured rapidly by dipstick and is of value in the diagnosis of diabetes insipidus and SIADH in the ED. Children with diabetes insipidus will typically have dilute urine with a specific gravity of less than 1.005 mOsm/kg.64 Those with SIADH, on the other hand, will have a concentrated urine, usually with a specific gravity above 1.030 mOsm/kg.
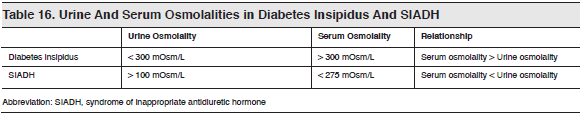
If available to the emergency clinician, measurements of urine and serum osmolalities are extremely valuable in diagnosing diabetes insipidus and SIADH. (See Table 16.)
In most cases, the diagnosis of diabetes insipidus will not be known at the time of presentation and will be considered only after initial resuscitation with isotonic saline. At this point, treat these patients the same as those being treated for hypernatremic dehydration. Although correction of hypernatremia is necessary to prevent long-term sequelae such as myelinolysis and cellular necrosis in the brain, correction must take place at a controlled rate.66 Overly zealous correction of hypernatremia results in cerebral edema, which can cause seizures, coma, permanent neurologic damage, and death.59,62,66-69 The correction rate for serum sodium should not exceed 0.5 mEq/L/hr when treating hypernatremia in children,70,71 and most studies have suggested correcting pediatric hypernatremia over a 48- to 72-hour period.66,72,73 Hypotonic fluid (0.2% sodium chloride or 0.45% sodium chloride, depending on the level of hypernatremia) can be used for this purpose, with no particular regimen having been proved to be superior to another. Given this fact, it is wise to discuss the particular treatment approach with a pediatric intensivist. Although desmopressin is the treatment of choice for diabetes insipidus, its use in the ED should be restricted to those children confirmed to have diabetes insipidus. This is another treatment option that should be used only after consultation with an appropriate subspecialist.
Like diabetes insipidus, SIADH in children is usually first considered once the initial laboratory results are known. Since hyponatremia is the main cause of morbidity in SIADH, treatment should be focused on correcting this electrolyte abnormality. Fluid restriction is the mainstay of therapy. In children with signs of cerebral edema, including altered mental status, seizures, or coma, more rapid correction is necessary. Although the use of 0.9% sodium chloride (154 mEq/L) would seem to be a reasonable option to correct severe hyponatremia, its use in SIADH specifically may, paradoxically, make the hyponatremia worse.63,74 Instead, 3% sodium chloride should be administered to increase the serum sodium level in children with symptomatic hyponatremia. Keep in mind that the key to using 3% sodium chloride is to raise the serum sodium level quickly enough to reverse the neurologic symptoms but not so quickly that it causes the dreaded complication of cerebral osmotic demyelination,75-77 which leads to various permanent neurologic disabilities and often death. The optimal rate at which to correct symptomatic hyponatremia is a matter of some controversy.63,78,79 Many authorities have suggested an infusion rate of 1 to 2 mL/kg/hr of 3% sodium chloride until neurologic symptoms improve or the serum sodium level exceeds 125 mEq/L.59,62,63,65,80 This should cause a rise in the serum sodium level of approximately 1 to 2 mEq/L/hr. Furosemide (1 mg/kg IV) can also be given to increase free water loss by the kidney.63 Check serum electrolytes every 2 hours in children who are being treated with 3% sodium chloride. Many authorities recommend not exceeding a serum sodium correction rate of more than 8 to 12 mEq/L/day.58,62,63,65,77,78,80,81
Admit any child in whom diabetes insipidus or SIADH is suspected. A fluid deprivation test may be necessary to confirm the diagnosis of diabetes insipidus. A search for other endocrinopathies may be necessary in both diabetes insipidus and SIADH. The close monitoring of fluid intake and output and of serum electrolytes is of more immediate importance in both conditions. Children with hemodynamic instability, any evidence of cerebral dysfunction, or a serum sodium level less than 125 mEq/L should be admitted to an intensive care setting.
A literature search was performed using Ovid MEDLINE® and PubMed. Keywords included acquired hypothyroidism, congenital hypothyroidism and hyperthyroidism, Graves’ disease, thyrotoxicosis, and thyroid storm. Similar searches were performed of the Cochrane Database of Systematic Reviews and the National Guideline Clearinghouse.
The majority of studies on pediatric hypothyroidism in recent decades have addressed congenital hypothyroidism, evaluating both programs that screen for this disorder and treatment options. Pediatric hyperthyroidism is a relatively rare disorder, with virtually all papers published on this topic being case series, and most recommendations about the treatment are individual or consensus opinions. Thyroid storm is exceptionally rare in children, with only a few case reports published; therefore, guidelines for managing this condition must be extrapolated from the adult literature.
Hypothyroidism is classified as primary (ie, resulting from a failure of the thyroid gland to produce thyroid hormones), secondary (ie, due to a lack of production of thyroid-stimulating hormone [TSH] by the pituitary), or tertiary (ie, caused by the failure of the hypothalamus to produce thyrotropin-releasing hormone [TRH]). Pediatric hypothyroidism can be congenital (when thyroid hormone production is inadequate at birth) or acquired (when it develops after the child has reached 6 months of age). Since thyroid hormones influence all aspects of normal development, including nervous system myelination, it is not surprising that untreated hypothyroidism is a significant cause of mental retardation worldwide.
Congenital hypothyroidism occurs in 1:3000 to 1:4000 newborns, and the female-to-male prevalence is 2:1.85 The majority of cases are due to dysgenetic or ectopic thyroid glands. The presenting symptoms are few and often nonspecific soon after birth, making it extremely difficult to diagnose clinically in the first few weeks of life.85 For this reason, newborn screening programs were developed in the 1970s and are now implemented in most industrialized nations. As with most screening programs, however, a small number of newborns who have congenital hypothyroidism are not detected.86,87
Acquired hypothyroidism starting after 2 years of age does not carry the risk of permanent mental retardation associated with congenital hypothyroidism. Since the routine addition of iodine to foods, the incidence of acquired hypothyroidism has decreased significantly in most developed countries. Autoimmune (Hashimoto) thyroiditis is the most common pediatric cause of acquired hypothyroidism in the United States.89 This illness occurs twice as often in females as males and usually presents in early to mid puberty. The incidence of Hashimoto thyroiditis during adolescence is approximately 1% to 2%.90 Other causes of acquired hypothyroidism are listed in Table 17.

A review of the key anatomic and physiologic features of the hypothalamic–pituitary–thyroid axis can be found in Figure 4.
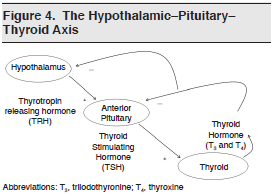
Hyperthyroidism occurs much less commonly than hypothyroidism in children. Thyrotoxicosis defines the clinical response to excessive amounts of thyroid hormones. Like hypothyroidism, pediatric hyperthyroidism can be divided into 2 main categories: congenital (neonatal) and acquired. Thyrotoxicosis factitia, hyperthyroidism caused by the ingestion of excessive amounts of thyroid hormone, has been reported with diet pill use among adolescents and accidental ingestions by toddlers.97 Other causes of pediatric hyperthyroidism are listed in Table 18.

Neonatal hyperthyroidism is caused by the transplacental passage of thyroid-stimulating immunoglobulins in pregnant women with Graves’ disease and rarely Hashimoto thyroiditis. It is estimated that less than 2% of infants born to mothers with Graves’ disease will manifest symptoms of hyperthyroidism.98 Neonatal thyrotoxicosis usually presents within a few days after birth but may be delayed for a week or more in infants born to mothers who are taking antithyroid medication.98,99 The duration of congenital hyperthyroidism is determined by the persistence of the maternal antibodies in the newborn, and the condition usually remits after 8 to 20 weeks.100 Although typically transient in nature, congenital hyperthyroidism may cause prolonged intellectual impairment.101
The vast majority of cases of acquired hyperthyroidism in developed countries are due to Graves’ disease, and it is estimated that only about 5% of all patients with this disease are diagnosed before age 18.102 Graves’ disease is caused by the production of antibodies directed against the thyroid hormone receptor on the thyroid gland as well as antigens in the orbital tissues. Given this fact, it is not surprising that up to 60% of children with Graves’ disease will have a family history of autoimmune thyroid disease.103 Pediatric Graves’ disease is rare in prepubertal children, peaks during adolescence, and affects girls approximately 4 times more often than it does boys.96 Uncontrolled hyperthyroidism in children may cause behavioral problems, emotional lability, accelerated bone maturation, and high-output cardiac failure.
Thyroid storm, also called thyrotoxic crisis, is a loosely defined clinical condition caused by excessive thyroid hormone release in a patient with preexisting hyperthyroidism. Patients present with tachycardia, fever, abdominal pain, emesis, and altered mental status ranging from agitation to seizures and coma. Although rare in children, this condition has a 20% mortality rate in adults. Thyroid storm typically results from a precipitating event such as surgery, severe infection, DKA, or trauma. Thyroid storm has also been reported in children after the discontinuation of antithyroid medication and after radioactive iodine therapy.104
Of all the symptoms and signs of hypothyroidism in children, lethargy in a neonate is the one situation most likely to lead to an ED visit. Although hypothyroidism is not common in the Western world, it should be considered a possibility in the differential diagnosis of a lethargic infant. (See Table 19).
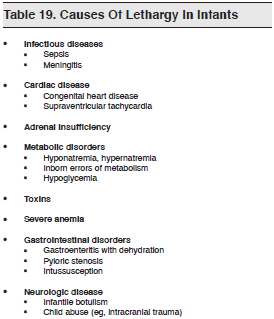
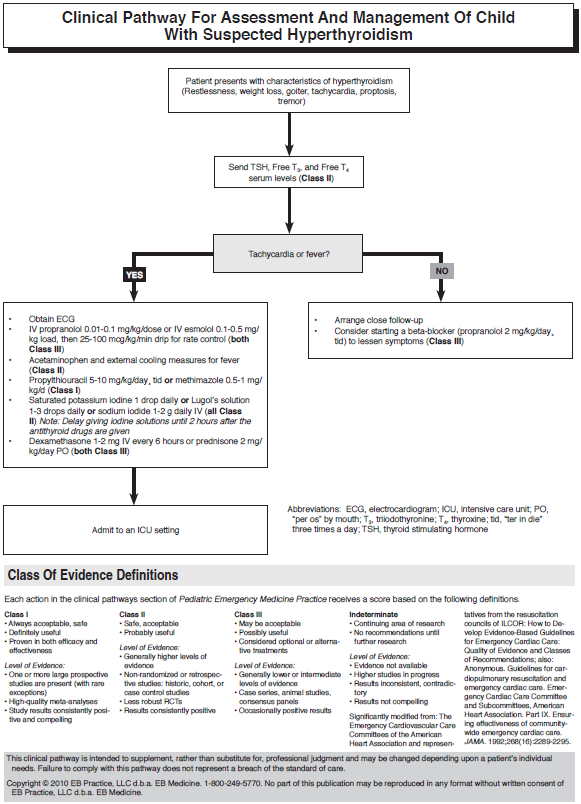
Many symptoms of pediatric hyperthyroidism are nonspecific and include weight loss, emotional lability, decreased school performance, frequent bowel movements, and feeling warm. One helpful finding, if present, is thyroid enlargement, which is present in most affected children. Be certain to check for thyromegaly in any child with unexplained tachycardia or tremor.
The predominant symptoms of thyroid storm (hyperpyrexia, mental status changes, and arrhythmias) can mimic several other illnesses, most notably CNS infections, sepsis, and anticholinergic or sympathomimetic ingestions. Children with thyroid storm may have a history of hyperthyroidism, but some rarely present with these symptoms because of a delay in the diagnosis. Review of systems in previously undiagnosed hyperthyroidism should be positive for the symptoms noted above, and the finding of a palpable goiter is further indication of the disease.
Children with hypothyroidism who present to the ED with lethargy should undergo finger-stick blood sugar testing and pulse oximetry; if opioid toxicity is suspected, IV or IM naloxone (0.1 mg/kg if < 20 kg and 2 mg if > 20 kg) should be administered.
Children with hyperthyroidism may require rapid treatment for hyperpyrexia and tachyarrhythmias, especially atrial fibrillation. Treat febrile children suspected of having hyperthyroidism with acetaminophen and with external cooling measures if fever is severe. Salicylates specifically should be avoided, since they displace thyroid hormone from binding proteins in the serum, resulting in an increase in free thyroid hormone.105 To achieve rate control in children with sinus tachycardia and atrial fibrillation, administer propranolol 0.01 to 0.1 mg/kg/dose IV or esmolol 0.1 to 0.5 mg/kg load IV, followed by an infusion of 25 to 100 mcg/kg/min. For children in whom beta-blockers are contraindicated, such as in severe asthma, diltiazem 0.25 mg/kg IV or digoxin 5 to 15 mcg/kg IV can be used for rate control in cases of atrial fibrillation.106
The common findings on history and physical examination in hypothyroidism are listed in Table 20.89,91-94 Since the vast majority of newborns with congenital hypothyroidism will not have clinical manifestations at birth, universal neonatal screening is essential to permit early intervention. If neonatal hypothyroidism is suspected, it is important to test thyroid function, since screening may not be done when infants are born at home or outside the United States. In addition, human error in screening labs or failure to follow up on abnormal results can occur.
The onset of symptoms in children with acquired hypothyroidism is often very subtle and may be apparent only to the primary care physician who follows a child’s maturation and development. The presence of goiter may be the only sign that would lead an emergency clinician to consider hypothyroidism. As with many autoimmune illnesses, thyroiditis may occur in association with other autoimmune disorders such as type I diabetes mellitus, celiac disease, or systemic lupus erythematosus. Be sure to ask about medications and a family history of thyroid disorders when considering a diagnosis of hypothyroidism in children. (See Table 17.)
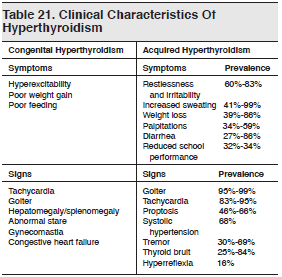
The characteristic history and physical findings in hyperthyroidism are summarized in Table 21.101,107-112 A thorough prenatal history may be the initial clue to the diagnosis of neonatal hyperthyroidism. The vast majority of infants with neonatal hyperthyroidism will be born to mothers with a history of current or prior treatment for Graves’ disease. Keep this fact in mind when evaluating a neonate for being “jittery” or “not feeding well.”
Many children diagnosed with acquired hyperthyroidism will, in retrospect, have had subtle symptoms for months or even years. Many of the early signs, such as restlessness, irritability, and decreased school performance, may be attributed to behavioral problems or even attention deficit–hyperactivity disorder.96 The most readily identifiable clinical signs of hyperthyroidism in children are goiter and unexplained tachycardia, both of which will be present in the majority of cases. The thyroid gland is usually diffusely and symmetrically enlarged. Roughly half of all children with hyperthyroidism will have proptosis, although this is typically not as pronounced as it is in adults.99,102
Unlike other manifestations of hyperthyroidism, thyroid storm is not a subtle clinical entity. Its defining features include fever as well as dysfunction of the cardiovascular, gastrointestinal, and central nervous systems.113 The temperature typically exceeds 38.9°C (102°F), and the pulse is usually above 140 beats/min, often with atrial fibrillation. Signs of congestive heart failure may be present. Vomiting and diarrhea are frequent symptoms, and the patient may be jaundiced. Signs of CNS dysfunction range from agitation to delirium and even coma. Some patients with thyroid storm may present with a seizure of new onset.
The laboratory abnormalities associated with severe, long-standing hypothyroidism include anemia, hyponatremia, hypercholesterolemia, and rarely hypoglycemia. The ideal screening tests for children with suspected hypothyroidism are TSH and free thyroxine (T4) levels.95,96 This combination allows for the identification of primary, secondary, and tertiary causes of hypothyroidism. Since the total T4 level can be altered by the availability of binding proteins in the serum, it is not as reliable as the free T4 level. Similarly, triiodothyronine (T3) is an unreliable indicator of hypothyroidism.
Measuring TSH is the single best screening test for hyperthyroidism, since this hormone will be undetectable in the vast majority of pediatric cases.114 Most authors also recommend obtaining a free T4 level and, if possible, a total or free T3 level.96,99,114-117 In a small minority of cases of hyperthyroidism, T3 will be elevated and free T4 will be normal. If a free T4 level cannot be obtained, a total T4 level can be used, even though it is not as reliable as free T4, as noted previously.
In those rare children in whom thyroid storm is suspected, additional testing will be required to detect multiorgan dysfunction. Electrolytes, serum glucose levels, and calcium levels should be measured, since these children will often have metabolic evidence of dehydration, hyperglycemia, and hypercalcemia owing to their hypermetabolic state. One should also rule out hepatic dysfunction, as indicated by elevations in liver transaminases, alkaline phosphatase, and bilirubin. An electrocardiogram is indicated to distinguish sinus tachycardia from other arrhythmias, most notably atrial fibrillation. Finally, some authors advocate urinalysis and a chest radiograph as a way of screening for a precipitating infection.118
The ultimate goal of treatment in pediatric hypothyroidism is to restore the euthyroid state as rapidly as possible. This is especially true for children under 2 years of age, in whom any delay in treatment will likely result in permanent neurologic deficits. On the rare occasion that pediatric hypothyroidism can be diagnosed and confirmed in the ED, treatment should begin immediately — ideally after consultation with a pediatric endocrinologist. A more likely scenario would be close follow-up with a pediatric endocrinologist or primary care physician to review screening results and initiate therapy. Treatment consists of daily oral levothyroxine and is easy and inexpensive. The recommended starting doses are based on age and are listed in Table 22.90,95
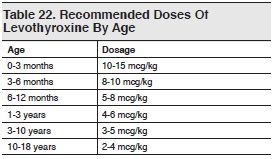
In pediatric hyperthyroidism, treatment must be individualized and should ideally be discussed with a pediatric endocrinologist. Other than in cases of suspected thyroid storm, specific treatment should be delayed until hyperthyroidism can be confirmed by the necessary laboratory tests (a rare occurrence in most EDs). Radioactive iodine therapy, thyroidectomy, and antithyroid drug therapy (ie, propylthiouracil [PTU] and methimazole [MMI] in the United States) are the main treatment modalities, with antithyroid drugs being the initial treatment of choice.115,117,119,120 Because of the recently recognized increase in the incidence of PTU-induced hepatotoxicity in children, MMI is now the antithyroid drug of choice for pediatric hyperthyroidism.121-123
The medications used in the treatment of pediatric hyperthyroidism are listed in Table 23. In those few children with symptomatic and confirmed hyperthyroidism, begin treatment with an antithyroid drug. These drugs block the synthesis of thyroid hormones but do not suppress the release of pre-existing hormones stored in the thyroid. To achieve the latter goal, an iodine-containing solution can be added to the treatment regimen; however, do not begin treatment with an iodine-containing solution until approximately 2 hours after the first dose of an antithyroid drug. This delay is necessary to assure that thyroid hormone synthesis is adequately blocked so that iodine cannot exert a potentially stimulating effect on hormone production. Iodine is usually reserved for the treatment of neonatal hyperthyroidism and thyroid storm. A beta-blocking agent can help to control tremors and tachycardia in those patients with more pronounced symptoms. Beta-blockers are typically required for only the first few weeks until thyroid hormone levels decline as a result of antithyroid medication.

Children believed to have thyroid storm will require treatment with an antithyroid drug, an iodine solution, a beta-blocker, and possibly a glucocorticoid. Glucocorticoids inhibit the conversion of T4 to T3 and will treat the relative adrenal suppression that is frequently present with thyroid storm.
Few, if any, children will require admission for the treatment of hypothyroidism, although close follow-up (ie, the next day) must be assured.
Since there are no guidelines regarding the treatment of pediatric hyperthyroidism, it would be wise to discuss the disposition of these children with their primary care physician or, ideally, a pediatric endocrinologist. Those children with mild symptoms and no tachycardia or fever may be discharged with close follow-up. Admit any child with suspected hyperthyroidism who has abnormal vital signs. Children with suspected thyroid storm are better managed in an intensive care setting.
Although endocrine emergencies other than DKA are uncommon in children, they are potentially life-threatening. These children typically present with nonspecific symptoms and many are critically ill. Attention to key findings on the history and physical examination can give the astute emergency clinician clues to the presence of these illnesses. Treatment of these children should ideally be coordinated with the appropriate pediatric subspecialist.
A rapid bedside glucose determination in children with polydipsia, polyuria, and clinical signs of dehydration quickly rules in or out diabetes mellitus as the cause. Risk Management Caveat: Finger-stick serum glucose measurements are not infallible and can be influenced by technique and user experience. This is another reason to order a chemistry panel in such cases in addition to evaluating serum electrolytes.
The dipstick urine specific gravity is a useful bedside test when one suspects diabetes insipidus (inappropriately dilute urine) or SIADH (inappropriately concentrated urine). Risk Management Caveat: Infants’ kidneys cannot concentrate urine as well as older children’s kidneys. Therefore, although specific gravity is a convenient early indicator of these conditions, it is neither 100% sensitive nor specific for them.
The septic-appearing 2-year-old did indeed have a left lower lobe pneumonia on chest x-ray and showed little improvement in her circulatory status after being given a 20 mL/kg bolus dose of normal saline. Finger-stick testing revealed a serum glucose of 52. Having recognized the picture of possible adrenal insufficiency in the face of sepsis, you drew and saved extra serum for confirmatory testing and gave a 2 mL/kg bolus of D25, 25 mg of IV hydrocortisone, another 20 mL/kg bolus of normal saline, and IV antibiotics. You discussed the case with the pediatric intensivist at the children’s hospital on the other side of town and arranged for a transfer. The child’s condition had improved significantly by the time the transport team arrived.

Evidence-based medicine requires a critical appraisal of the literature based upon study methodology and number of subjects. Not all references are equally robust. The findings of a large, prospective, randomized, and blinded trial should carry more weight than a case report.
To help the reader judge the strength of each reference, pertinent information about the study, such as the type of study and the number of patients in the study, will be included in bold type following the reference, where available. In addition, the most informative references cited in this paper, as determined by the authors, will be noted by an asterisk (*) next to the number of the reference.
Wesley Eilbert
April 1, 2011
Pediatric Emergency Medicine Practice • CONTINUE READING
Access every issue, our complete clinical pathway library, and earn up to 190 CME credits with an annual subscription.
Stay current with a new Pediatric Emergency Medicine Practice issue every month, plus unlimited access to our complete issue library, all Interactive Clinical Pathways, and up to 190 CME credits.
Accredited By





Our Partners


 678-366-7933
678-366-7933